


Quick Research
Generate reliable direction feasibility study reports for your R&D in just a few steps.

Technical Q&A
Discover and master advanced knowledge NOW. Basics, ideas, possibilities, all at once.

Find Solutions
As an expert in R&D theories, this can generate solutions to your technical problems instantly.

Evaluate Feasibility
Analyze your overall solution with one click, know your potential R&D risks in advance.

Monitor Landscape
Get weekly tech updates, stay abreast of the latest tech innovations and key insights.
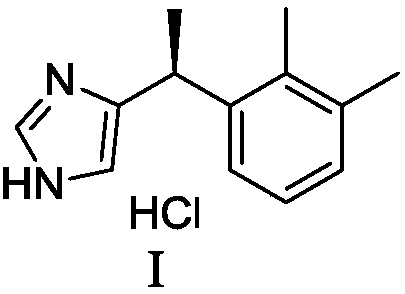
Preparation method of dexmedetomidine hydrochloride and its intermediate
A technology for dexmedetomidine and intermediates, applied in the field of preparation of dexmedetomidine hydrochloride and its intermediates, can solve the problems of long steps, low molar yield, unsuitable for industrial production, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


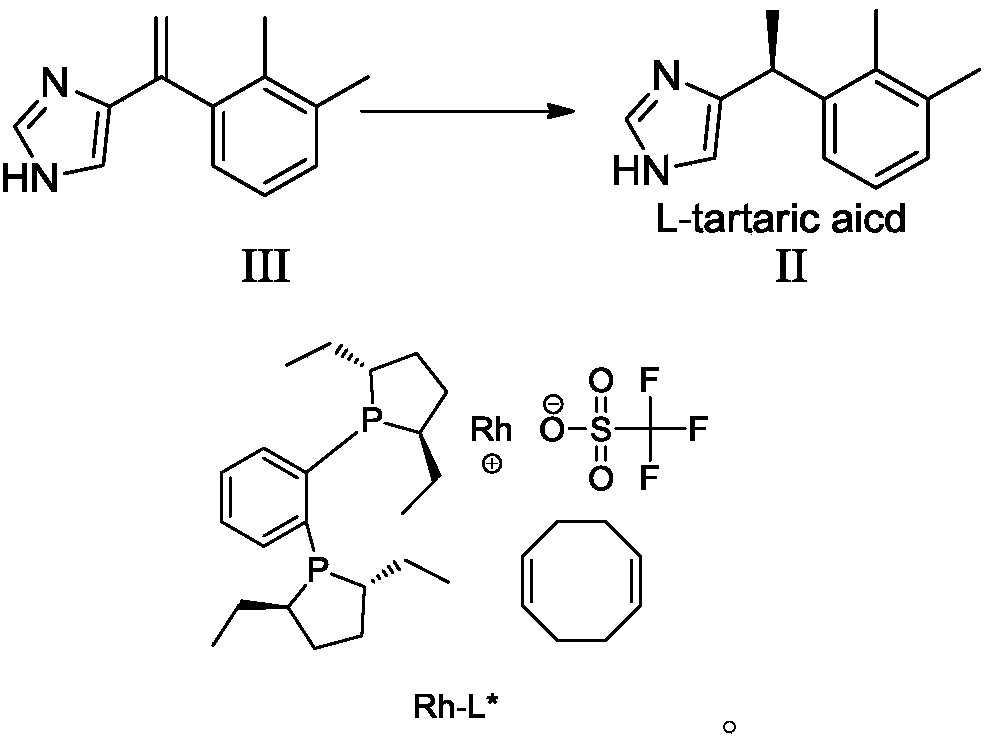
Examples
Embodiment 1
[0106] Example 1: Preparation of trimethylsilyl-2,3-dimethylphenylacetylene
[0107]
[0108]Under the protection of argon, 55.0 g of 2,3-dimethylbromobenzene was added to 440 mL of tetrahydrofuran, followed by 1.05 g of bistriphenylphosphine palladium dichloride, 62.0 g of cuprous iodide, and 60.0 g of triethylamine. After vacuum degassing and argon replacement, heat to 45°C, slowly add a solution of 35.0 g of trimethylsilylacetylene in 140 mL of tetrahydrofuran dropwise, and react for 8 to 10 hours. After cooling, filter, add mass concentration and be 10% ammonium chloride aqueous solution (the described mass concentration refers to the quality of ammonium chloride accounts for the percentage of ammonium chloride aqueous solution total mass) 100mL and water 100mL, extract with n-heptane 200mL Twice, merging organic phase with mass concentration is that 10% sodium bicarbonate aqueous solution (described mass concentration refers to the percentage that the quality of sodium...
Embodiment 2
[0109] Example 2: Preparation of trimethylsilyl-2,3-dimethylphenylacetylene
[0110]
[0111] Under the protection of argon, 55.0 g of 2,3-dimethylbromobenzene was added to 825 mL of 2-methyltetrahydrofuran, and then 2.19 g of dichlorobis(tricyclohexylsulfone)palladium, 84.5 g of cuprous iodide, and Isopropylethylamine 115g. After vacuum degassing and argon replacement, heat to 30°C, slowly add dropwise a solution of 43.8 g of trimethylsilylacetylene in 140 mL of 2-methyltetrahydrofuran, and react for 13 to 15 hours. After cooling, filter, add mass concentration and be 10% ammonium chloride aqueous solution (the described mass concentration refers to the quality of ammonium chloride accounts for the percentage of ammonium chloride aqueous solution total mass) 100mL and water 100mL, extract with n-heptane 200mL Twice, merging organic phase with mass concentration is that 10% sodium bicarbonate aqueous solution (described mass concentration refers to the percentage that the ...
Embodiment 3
[0112] Example 3: Preparation of trimethylsilyl-2,3-dimethylphenylacetylene
[0113]
[0114] Under argon protection, add 55.0 g of 2,3-dimethylbromobenzene to 303 mL of 1,4-dioxane, and then add [1,1'-bis(diphenylphosphino)ferrocene] 0.59 g of palladium dichloride, 45.0 g of cuprous iodide, and 71.0 g of tri-n-butylamine. After vacuum degassing and argon replacement, heat to 60°C, slowly add dropwise a solution of 30.6 g of trimethylsilylacetylene in 140 mL of 1,4-dioxane, and react for 5 to 7 hours. After cooling, filter, add mass concentration and be 10% ammonium chloride aqueous solution (the described mass concentration refers to the quality of ammonium chloride accounts for the percentage of ammonium chloride aqueous solution total mass) 1000mL and water 1000mL, extract with n-heptane 600mL Twice, merging organic phase with mass concentration is that 10% sodium bicarbonate aqueous solution (described mass concentration refers to the percentage that the quality of sod...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D Engineer
- R&D Manager
- IP Professional
- Industry Leading Data Capabilities
- Powerful AI technology
- Patent DNA Extraction
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2024 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com