


Quick Research
Generate reliable direction feasibility study reports for your R&D in just a few steps.

Technical Q&A
Discover and master advanced knowledge NOW. Basics, ideas, possibilities, all at once.

Find Solutions
As an expert in R&D theories, this can generate solutions to your technical problems instantly.

Evaluate Feasibility
Analyze your overall solution with one click, know your potential R&D risks in advance.

Monitor Landscape
Get weekly tech updates, stay abreast of the latest tech innovations and key insights.
Green synthesis method of antiviral drug intermediate
A synthesis method and compound technology, applied in organic chemistry methods, chemical instruments and methods, organic compounds/hydrides/coordination complex catalysts, etc. The effect of short, low reaction cost and easy operation
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

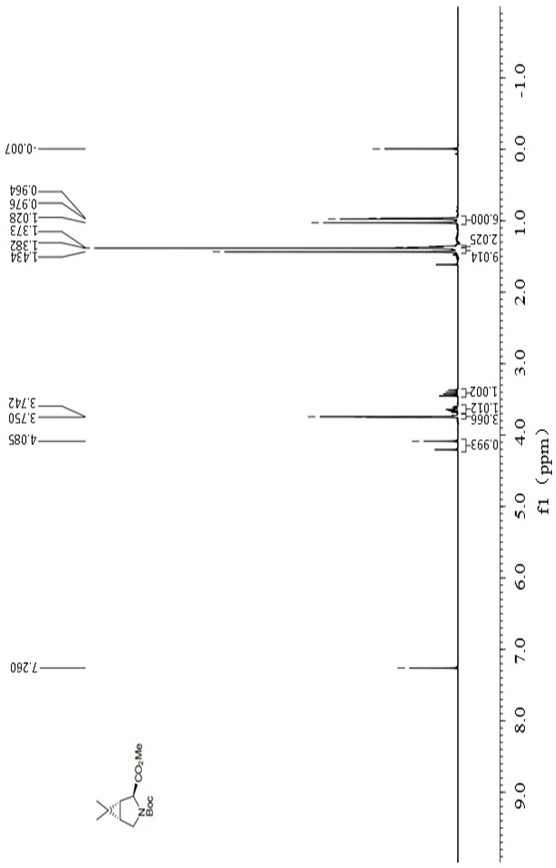
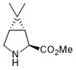
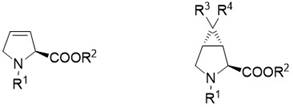
Examples
Embodiment 1
[0053] Example 1 Preparation of catalyst
[0054] (1) Preparation of ligands
[0055] 2,6-diacetylpyridine and optionally substituted aniline, in an organic solvent (such as toluene), the reaction is heated to reflux for 24 hours, and concentrated under reduced pressure to obtain a solid, which can be selectively purified to obtain the ligand shown in the following formula III compound;
[0056]
[0057] Formula III
[0058] R 5 Selected from methyl, ethyl, n-propyl, isopropyl or n-butyl.
[0059] For example the preparation of the catalyst 2,6-bis[1-[2-(ethylphenyl)imino]ethyl]pyridine may include:
[0060]
[0061] 2,6-Diacetylpyridine (16.3g, 0.1mol) and 2-ethylaniline (24.3g, 0.2mol) were successively added to a three-necked flask containing 200mL of toluene, and a catalytic amount of p-toluenesulfonic acid (1.72g) was added. , 0.01mol), stirred and mixed, and the reaction solution was heated to reflux for 24h. The reaction solution was directly concentrated to...
Embodiment 2
[0067]
[0068] 1. Preparation of methyl ligand L1 with reference to Example 1, taking methyl ligand L1 and CoBr 2 The catalyst solid is prepared;
[0069] 2. Add 24.6 g of the above catalyst solid and 100 g of the substrate N-Boc-3,4-dehydro-L-proline, dissolve it in a three-necked flask with 1000 mL of anhydrous THF (tetrahydrofuran), replace it with nitrogen, and do a good job of Oxygen operation, mechanical stirring;
[0070] 3. Add 57.2 g of zinc powder into the reaction flask at room temperature, and stir mechanically;
[0071] 4. Stir at room temperature for 20 minutes, at which time the reduced cobalt catalyst in the solution shows a dark purple color;
[0072] 5. Add 235g of 2,2-dibromopropane again, and continue to stir the reaction for 3 hours at room temperature;
[0073] 6. After the reaction finishes, the reaction solution is directly concentrated under reduced pressure to obtain a crude product;
[0074] 7. the crude product is dispersed by adding 10 time...
Embodiment 3
[0076]
[0077] 1. Preparation of methyl ligand L1 with reference to Example 1, taking methyl ligand L1 and CoBr 2 The catalyst solid is prepared;
[0078] 2. Add the above catalyst solid (246 mg, 0.44 mmol), the substrate N-Boc-3,4-dehydro-L-proline (1.0 g, 4.4 mmol), and dissolve it in 25 mL of 10 mL of anhydrous THF. In the three-necked bottle, nitrogen is replaced, anaerobic operation is performed, and magnetic stirring is performed;
[0079] 3. Add zinc powder (572 mg, 8.8 mmol) to the reaction flask at room temperature and stir magnetically;
[0080] 4. Stir at room temperature for 15 minutes, at which time the reduced cobalt catalyst in the solution shows a dark purple color;
[0081] 5. Add 2,2-dibromopropane (2.4 g, 8.8 mmol) again, and continue to stir the reaction for 1 hour at room temperature;
[0082] 6. After the reaction finishes, the reaction solution is directly concentrated under reduced pressure to obtain a crude product;
[0083] 7. The crude produc...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D Engineer
- R&D Manager
- IP Professional
- Industry Leading Data Capabilities
- Powerful AI technology
- Patent DNA Extraction
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2024 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com