Genetic diagnosis using multiple sequence variant analysis
a gene and multiple sequence technology, applied in the field of nucleic acid-based genetic analysis, can solve the problems of complex structure, shaped, and not simple functions of ld, and achieve the effects of improving the accuracy of results and reducing the difficulty of diagnosis
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image



Examples
example 1
Intraspecies SPC Map of the Sh2 Locus of Maize
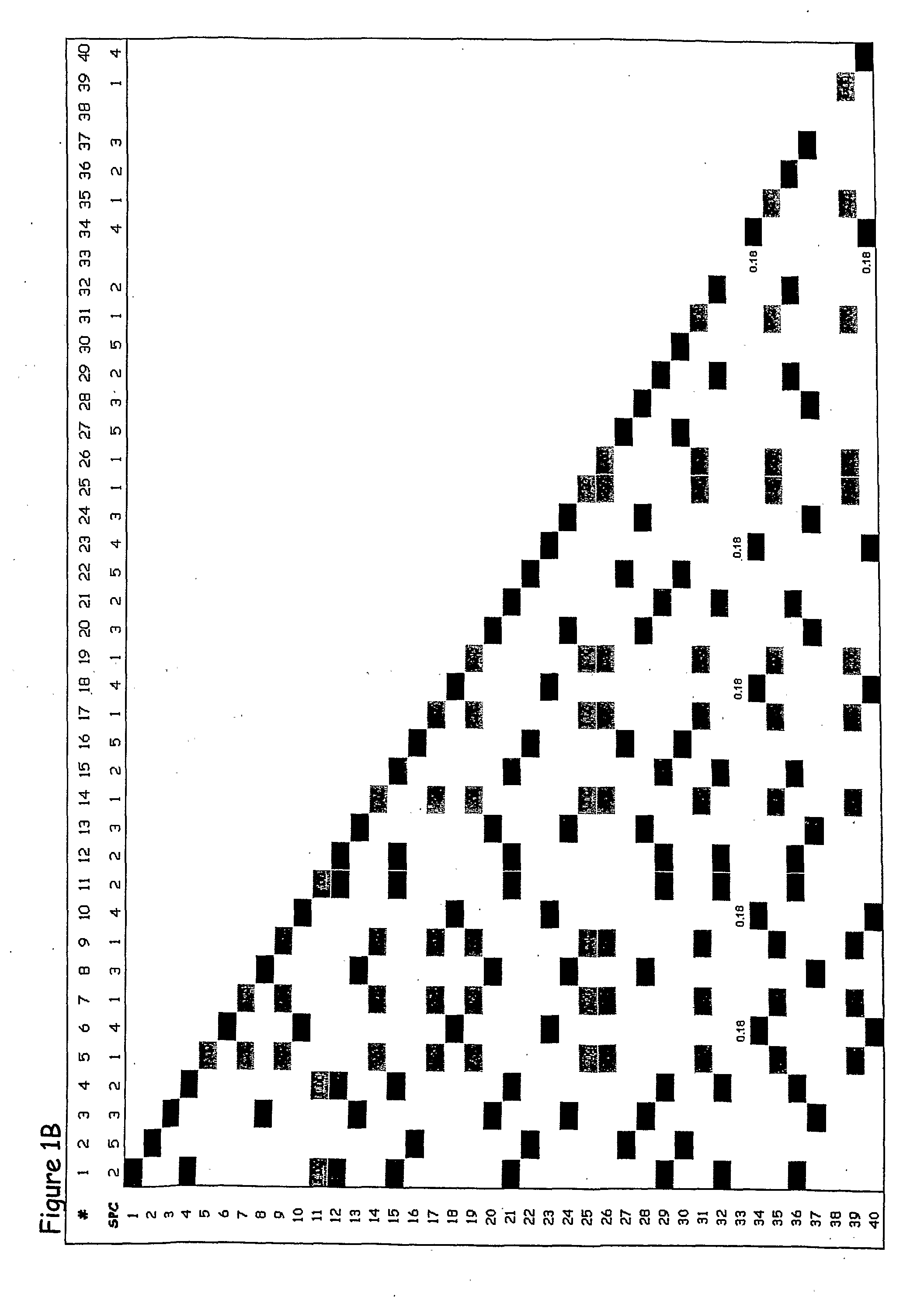
[0321]The present example provides proof of concept that the methods of the present invention can be used to generate an SPC map of a complete gene locus that has been sequenced in a number of individuals of a particular species. Many studies on the genetic diversity of specific genes have been conducted in a broad range of plant and animal species, and these sequences are publicly available from GenBank (http: / / www.ncbi.nlm.nih.gov). In most of these studies relatively short gene segments, less than 1000 bp, have been sequenced and only in a few studies have complete genes been sequenced. From the available complete or near complete gene sequences available in GenBank, the shrunken2 (sh2) locus from maize was chosen to exemplify the different aspects of the invention. The published shrunken2 locus sequences from 32 maize cultivars (Zea mays subsp. mays) comprise a region of 7050 bp containing the promoter and the coding region of the sh...
example 2
Intraspecies SPC Map of the sh1 Locus of Maize
[0329]The present example provides proof of concept that the methods of the present invention can be used to generate an SPC map of a complete gene in which extensive recombination has occurred. This example presents an analysis of the polymorphic sites in the shrunken1 (sh1) locus from maize to exemplify further aspects of the invention. The published shrunken1 locus sequences from 32 maize cultivars (Zea mays subsp. mays) comprise a region of 6590 bp containing the promoter and the coding region of the sh2 gene [Whitt et al., Proc. Natl. Acad. Sci. USA 99: 12959-12962, 2002].
[0330]The sequences for this analysis were retrieved from GenBank (http: / / www.ncbi.nlm.nih.gov) accession numbers AF544100-AF544131. The sequences were aligned to generate a genetic variation table as described in detail in Example 1. The genetic variation table of the sh1 gene comprises 418 polymorphic sites. Because of this very large number of polymorphic sites,...
example 3
Intraspecies SPC Map of the Y1 Locus of Maize
[0333]The present example provides proof of concept that the method of the present invention can be used to generate an SPC map of a locus in which several historical recombination events have occurred. This example presents an analysis of the polymorphisms in the Y1 phytoene synthase locus of maize to exemplify further aspects of the invention. The Y1 phytoene synthase gene, which is involved in endosperm color, was sequenced in 75 maize inbred lines [Palaisa et al., The Plant cell 15: 1795-1806, 2003], comprising 41 orange / yellow endosperm lines and 32 white endosperm lines.
[0334]The sequences for this analysis were retrieved from GenBank (http: / / www.ncbi.nlm.nih.gov) accession numbers AY296260-AY296483 and AY300233-AY300529. The sequences comprise 7 different segments from a region of 6000 bp containing the promoter and the coding region of the Y1 phytoene synthase gene. The individual sequences were aligned to generate 7 genetic varia...
PUM
| Property | Measurement | Unit |
|---|---|---|
| nucleic acid sequence | aaaaa | aaaaa |
| nucleic acid | aaaaa | aaaaa |
| differential nucleic acid analysis | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com