Preparation method of amiodarone hydrochloride intermittent
A technology for amiodarone hydrochloride and intermediates, which is applied in the preparation of amiodarone hydrochloride intermediates for antiarrhythmic drugs and the field of preparation of 2-butyl-3-benzofuran, which can solve the problems of low reaction yield and cyclization Low reaction conversion rate, cost increase and other problems, to achieve the effect of simple operation and high reaction conversion rate
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


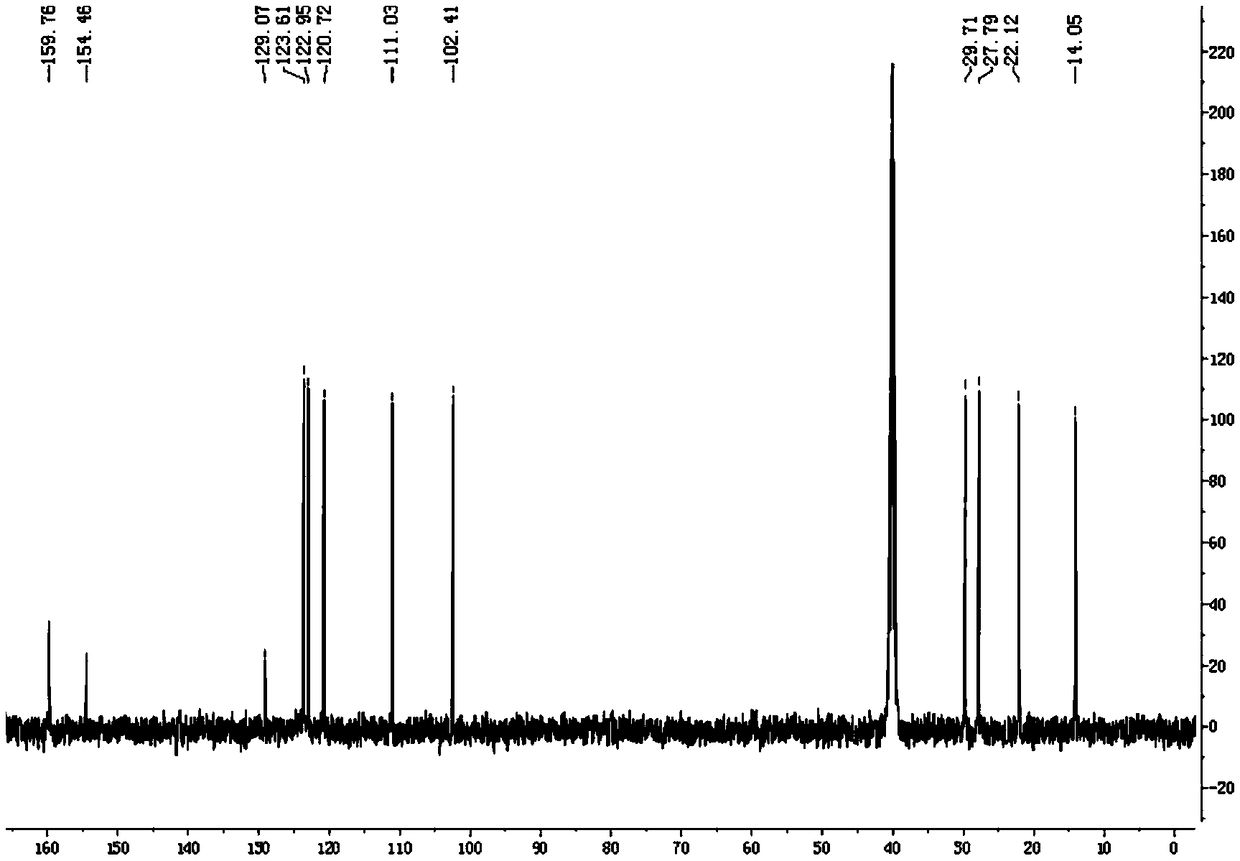
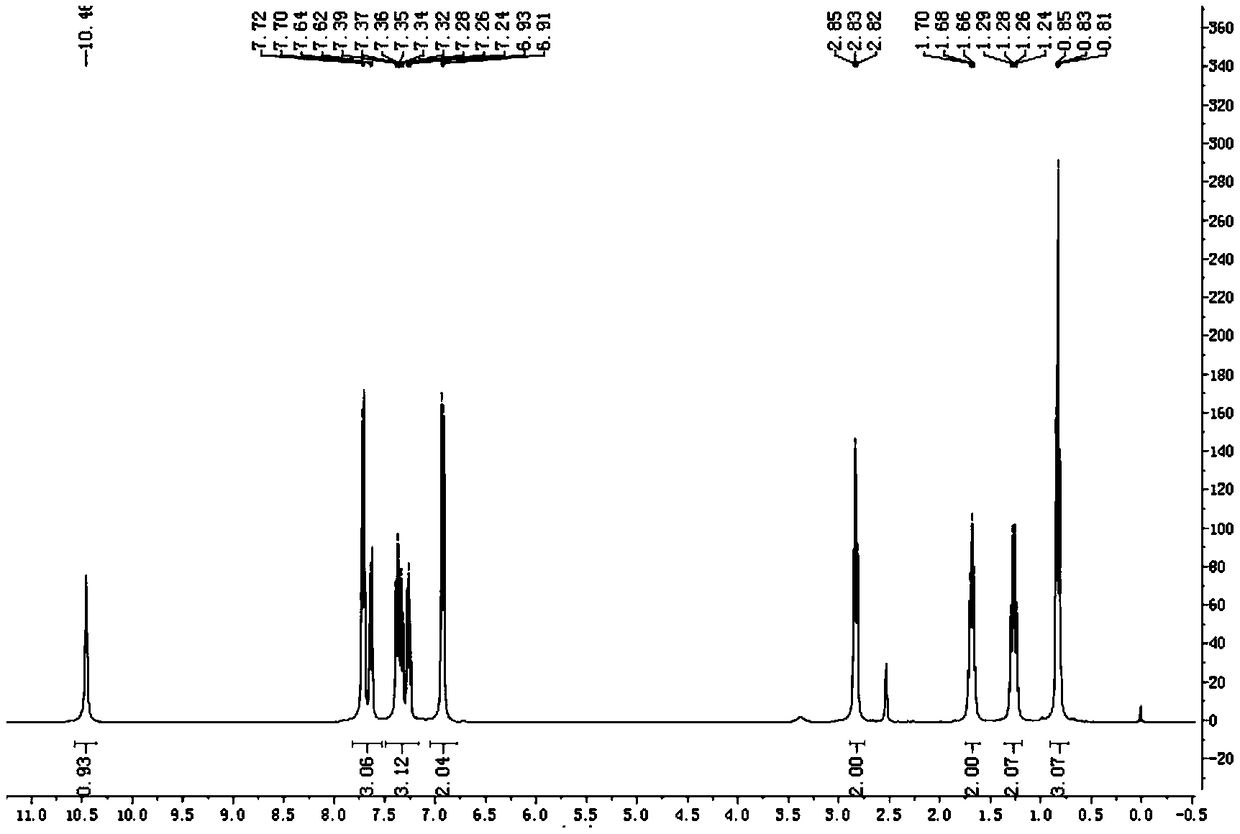
Examples
Embodiment 1
[0054]15.00kg (122.8mol, 1eq) of compound 1 and 30.82kg (147.4mol, 1.2eq) of compound 2 were added to 120.00kg (8w / w) of ethyl acetate, and 24.00kg (73.7mol, 0.6eq) of cesium carbonate was added, 1.00kg (2.5mol, 0.02eq) of methyl trioctyl ammonium chloride, stirred and heated to 75-85°C, reacted for 1-2 hours. Suction filtration after the reaction was completed, the filtrate was washed with 60.00kg (4w / w) purified water, and the organic phase was concentrated to dryness at 40°C under reduced pressure to obtain 30.21kg (120.7mol) of compound 3 with a yield of about 98.3%.
[0055] Add 30.00kg (120.0mol, 1eq) of compound 3 to 14.40kg (360.0mol, 3eq) of 150.00kg (5w / w) aqueous solution of sodium hydroxide, and stir at 20-30°C. After the reaction, add 1N dilute hydrochloric acid to adjust the pH to 4, add 30.00kg (1w / w) ethyl acetate for extraction, add 30.00kg (1w / w) purified water to the organic phase to wash once, add 30.00kg (1w / w) to the organic phase Wash with saturated sod...
Embodiment 2
[0063] 15.00kg (122.8mol, 1eq) of compound 1 and 30.82kg (147.4mol, 1.2eq) of compound 2 were added to 120.00kg (8w / w) of ethyl acetate, and 10.19kg (73.7mol, 0.6eq) of potassium carbonate was added, 1.00kg (2.5mol, 0.02eq) of methyl trioctyl ammonium chloride, stirred and heated to 75-85°C, reacted for 1-2 hours. Suction filtration after the reaction was completed, the filtrate was washed with 60.00 kg (4w / w) purified water, and the organic phase was concentrated to dryness at 40°C under reduced pressure. 26.21kg (104.7mol) of compound 3 were obtained, the yield was about 85.3%.
[0064] Add 26.00kg (103.9mol, 1eq) of compound 3 to 12.47kg (311.7mol, 3eq) of sodium hydroxide in 130.00kg (5w / w) methanol solution, and stir at 20-30°C. After the reaction was completed, it was concentrated to dryness under reduced pressure at 40°C. Add 130.00kg (5w / w) aqueous solution to dissolve, add 1N dilute hydrochloric acid to adjust the pH to 4, add 26.00kg (1w / w) ethyl acetate for extrac...
Embodiment 3
[0071]15.00kg (122.8mol, 1eq) of compound 1 and 30.82kg (147.4mol, 1.2eq) of compound 2 were added to 120.00kg (8w / w) of toluene, and 24.00kg (73.7mol, 0.6eq) of cesium carbonate was added, 1.00kg (2.5mol, 0.02eq) methyl trioctyl ammonium chloride, stir and heat up to 75-85°C, react for 1-2 hours. Suction filtration after the reaction was completed, the filtrate was washed with 60.00 kg (4w / w) purified water, and the organic phase was concentrated to dryness under reduced pressure at 40°C. 30.36 kg (121.3 mol) of compound 3 were obtained, with a yield of about 98.8%.
[0072] Add 30.00kg (120.0mol, 1eq) of compound 3 to 20.20kg (360.0mol, 3eq) of potassium hydroxide in 150.00kg (5w / w) aqueous solution, and stir at 20-30°C. After the reaction, add 1N dilute hydrochloric acid to adjust the pH to 4, add 30.00kg (1w / w) ethyl acetate for extraction, add 30.00kg (1w / w) purified water to the organic phase to wash once, add 30.00kg (1w / w) to the organic phase Wash with saturated sod...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com