


Quick Research
Generate reliable direction feasibility study reports for your R&D in just a few steps.

Technical Q&A
Discover and master advanced knowledge NOW. Basics, ideas, possibilities, all at once.

Find Solutions
As an expert in R&D theories, this can generate solutions to your technical problems instantly.

Evaluate Feasibility
Analyze your overall solution with one click, know your potential R&D risks in advance.

Monitor Landscape
Get weekly tech updates, stay abreast of the latest tech innovations and key insights.
Method for synthesizing key intermediate of antitumour medicament Romidepsi
A technology of romidistatide and anti-tumor drugs, which is applied in the field of drug synthesis, and can solve the problems of expensive reagents, low yield, and many reaction steps
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

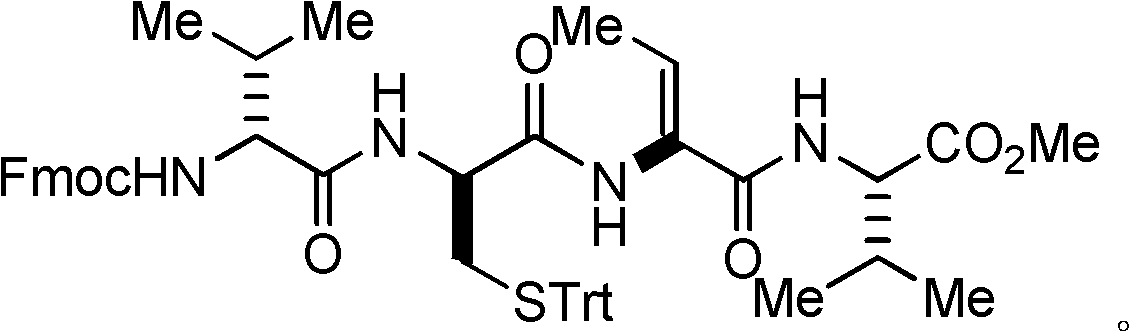
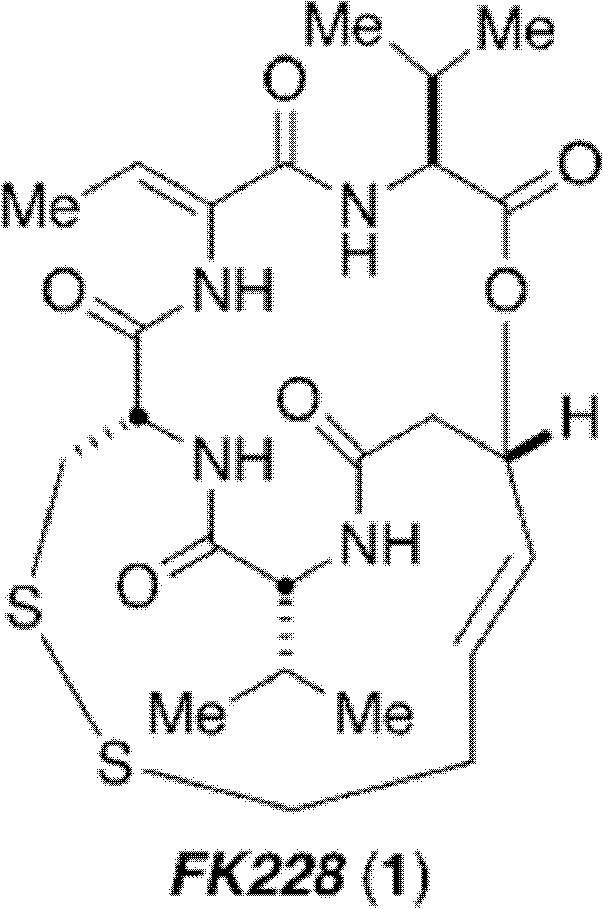
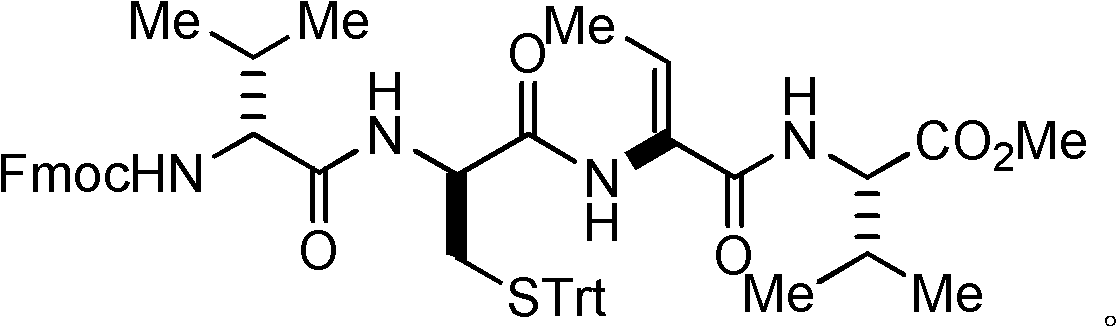
Examples
Embodiment 1
[0050] Example 1: Preparation I of dipeptide protected by fluorenyl moxycarbonyl group.
[0051] L-valine methyl ester hydrochloride (L-Val-OMe·HCl, 168 mg, 1 mmol) and fluorenylmethoxycarbonyl-L-threonine (N-Fmoc-L-Thr, 409 mg, 1.2 millimoles) into a 25 milliliter flask, and at room temperature, 15 milliliters of acetonitrile was added for dissolution, after which the flask was evacuated with an oil pump and flushed with nitrogen several times, so that the flask was filled with nitrogen. Turn on the magnetic stirrer and stir slowly at room temperature, and then add benzotriazol-1-yl-oxytripyrrolidinyl phosphorus hexafluorophosphate (624.4 mg, 1.2 mmol) and basic catalyst nitrogen, nitrogen diisopropyl Diethylamine (DIEA) (0.52 ml, 3 mmol) was added into the flask, and then the magnetic stirrer was accelerated for stirring. After reacting at room temperature for 30 minutes, the stirring was stopped to end the reaction. Concentrate under reduced pressure with a rotary evaporat...
Embodiment 2
[0055] Example 2: Preparation II of fluorenylmethoxycarbonyl-protected dipeptides.
[0056] L-valine methyl ester hydrochloride (L-Val-OMe·HCl, 168 mg, 1 mmol) and fluorenylmethoxycarbonyl-L-threonine (N-Fmoc-L-Thr, 682.8 mg, 2 mmol) was added into a 25 ml flask, and 20 ml of dichloromethane was added to dissolve at room temperature, after which the flask was evacuated with an oil pump and flushed with nitrogen several times, so that the flask was filled with nitrogen. Turn on the magnetic stirrer and stir slowly at room temperature, and then add benzotriazol-1-yl-oxytripyrrolidinyl phosphorus hexafluorophosphate (1040.6 mg, 2.0 mmol) and basic catalyst nitrogen, nitrogen diisopropyl Diethylamine (DIEA) (1.04 ml, 6 mmol) was added into the flask, and then the magnetic stirrer was accelerated for stirring. After reacting at room temperature for 120 minutes, the stirring was stopped to end the reaction. Concentrate under reduced pressure with a rotary evaporator to remove the s...
Embodiment 3
[0057] Embodiment 3: Preparation I of dipeptide.
[0058] The obtained dipeptide (obtained in Example 1, 460 mg, 0.98 mmol) protected by the fluorenyl moxycarbonyl group was added to a 50 ml flask, and 20 ml of acetonitrile was added to dissolve at room temperature, after which the flask was evacuated with an oil pump and flushed with nitrogen. Fill the flask with nitrogen several times. Turn on the magnetic stirrer to stir at room temperature, then add diethylamine (1 ml, 9.8 mmol) dropwise into the flask at 0° C., then stir at room temperature for 3 hours, then stop stirring to end the reaction. Concentrate under reduced pressure with a rotary evaporator to remove the solvent to obtain a crude dipeptide product, which can be directly used in the next reaction without further purification.
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D Engineer
- R&D Manager
- IP Professional
- Industry Leading Data Capabilities
- Powerful AI technology
- Patent DNA Extraction
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2024 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com