Construction method and application of high-throughout sequencing library
A sequencing library, high-throughput technology, used in the methylation detection of specific regions of the genome, the construction of high-throughput sequencing libraries, the kit for high-throughput sequencing libraries in specific regions of the genome, and the field of DNA methylation detection. issues to be improved
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

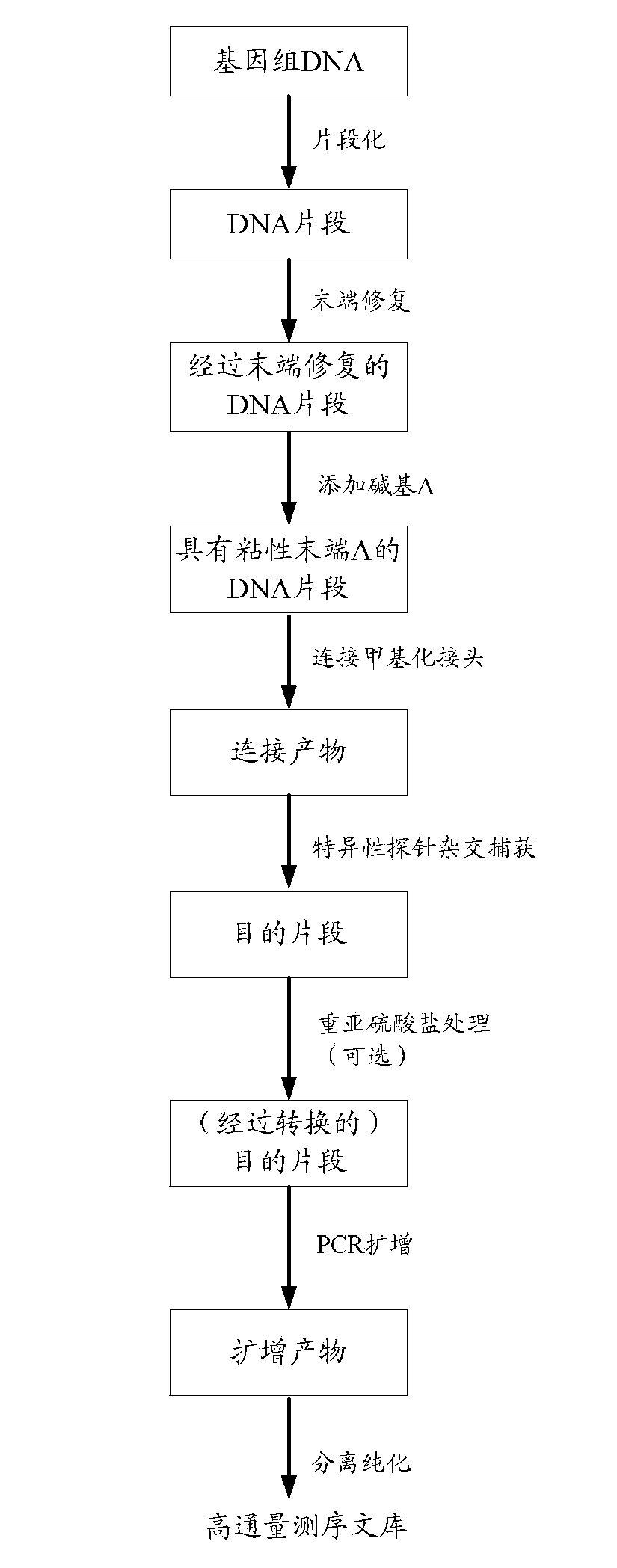

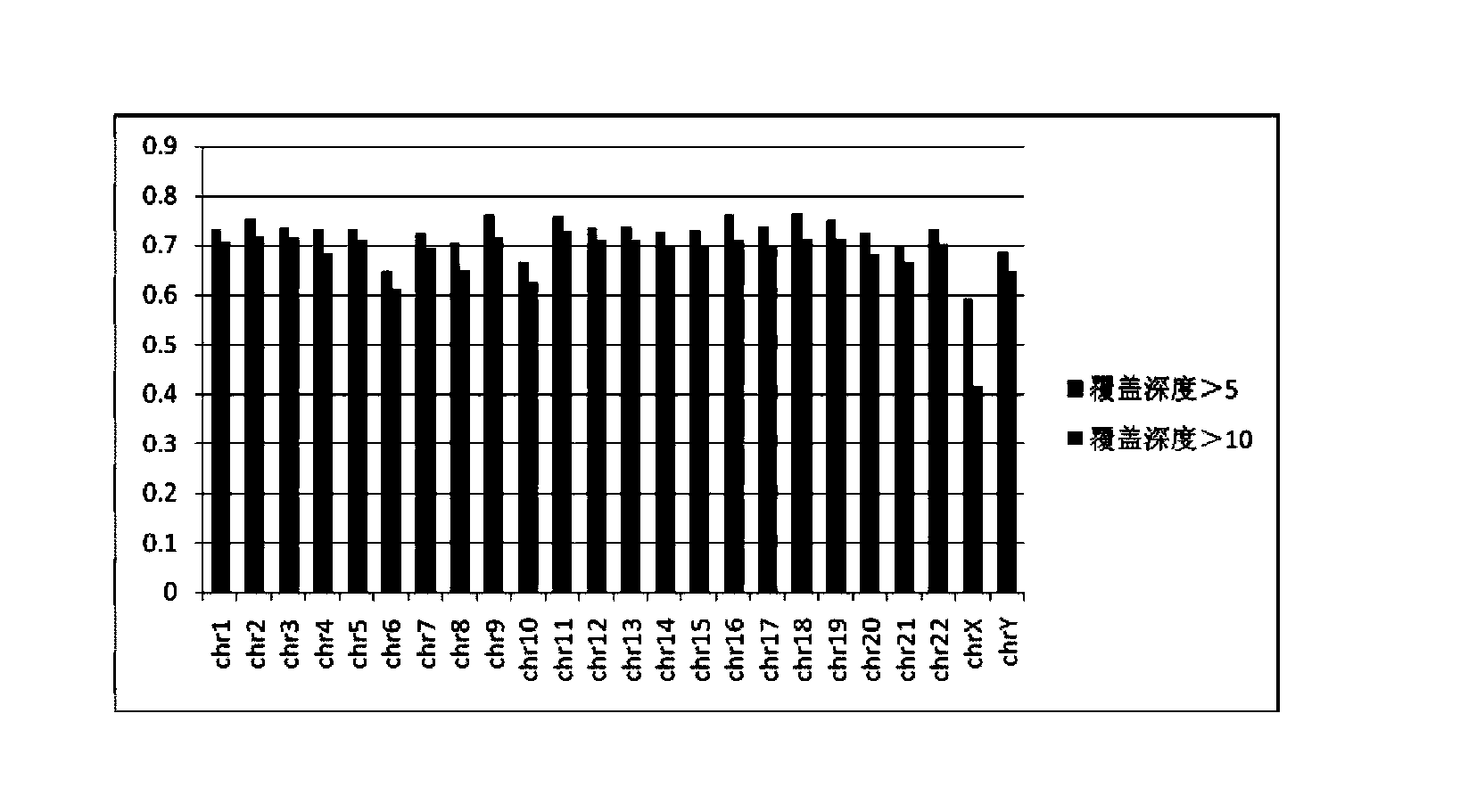
Examples
Embodiment 1
[0086] In this embodiment, 2 μg of human peripheral blood mononuclear cell genomic DNA was used as a sample, and the implementation was carried out according to the following steps.
[0087] 1. Genomic DNA fragmentation:
[0088] Use the covaris-S2 fragmentation instrument to fragment the genomic DNA of the sample according to the parameters set in the table below, so as to obtain DNA fragments.
[0089] processing 1
Load ratio (%)
10
[0090]
intensity
5
cycle / pulse
200
time(s)
50
time(s)
0
time(s)
0
Process 4
time(s)
0
cycle
3
[0091] The obtained DNA fragments are detected by electrophoresis, and the main band of the DNA fragments is required to be concentrated between 150-300bp, without protein and RNA contamination. Using QIAquick PCR Purification Kit (Qiagen) or magnetic bead purification, the qualifi...
Embodiment 2
[0189] Using the Hiseq2000 sequencer, the high-throughput sequencing library of the specific genome region of the sample constructed in Example 1 was sequenced according to the read length of 90 bases at both ends, so as to obtain the sequencing results.
[0190] After the above-mentioned sequencing, the raw data is directly obtained, and the above-mentioned sequencing results can be obtained by performing basic analysis on the raw data, wherein the basic analysis process includes the following main steps: First, distinguish different samples through the sequence tags on the adapters or PCR primers library data; then, decontamination, joint removal, and low-quality filtering are performed on the raw data obtained by sequencing; finally, base conversion is performed on the previously processed data, specifically, all Cs of the positive strand are converted into Ts, All the Gs of the complementary strands were converted into A, thus, the sequencing results of the high-throughput ...
Embodiment 3
[0203] Using Yanhuang cell line samples (Jun Wang et al.2008), repeat Example 1, except that the gene regions known to have methylation sites used to design specific probes are the codes of the genes listed in Table I Region and promoter region (867 genes in total after merging repeated genes), designed by eArray system, prepared by Agilent, the length of the probe is 12mer. Additionally, no bisulfite treatment step is required for resequencing and unmethylated sequencing libraries.
[0204] Using mixed index sequencing, the read length is 49bp, the index length is 6bp, the number of off-machine sequence fragments is 2.67Mb pairs, and the test data output is about 240Mb. Using the bwa alignment program, the sequenced fragments filtered for low-quality and contaminated adapters were aligned to the whole human genome. And a preliminary analysis of the comparison results was made.
[0205] Test results :
[0206] Table 3 shows the specific total amount of data off-machine of...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com