Method for production of furanose derivative
A manufacturing method and compound technology, applied in the direction of sugar derivatives, sugar derivatives, esterified saccharides, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

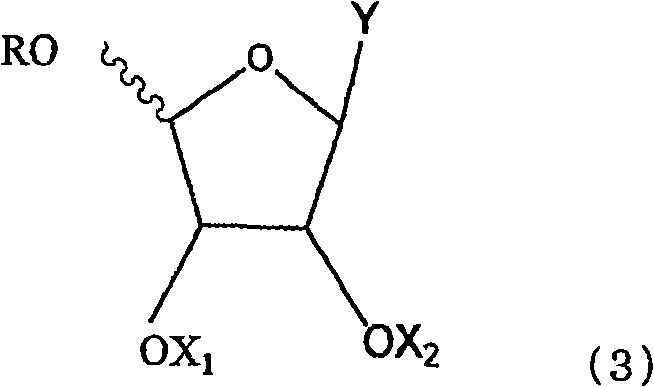
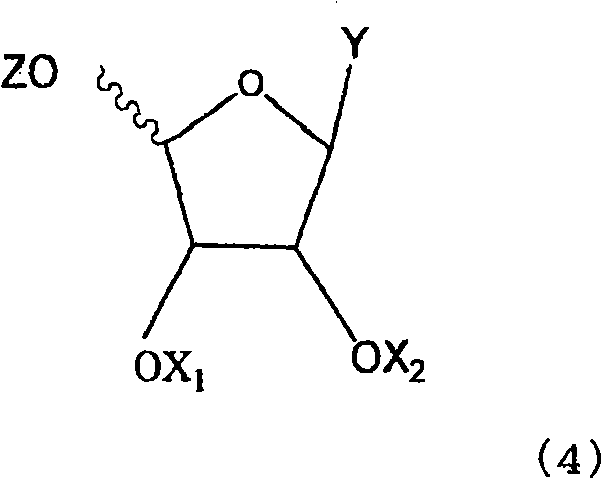
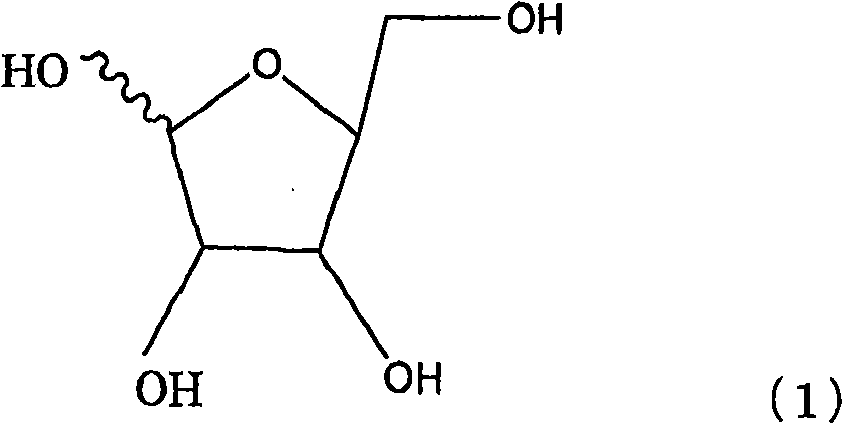
Examples
Embodiment A1
[0108] (1) Synthesis of 1-O-methyl-L-ribofuranose
[0109] L-ribose (20.0 g, 132 mmol) and methanol (226 g) were added to a 1000 mL flask, and concentrated sulfuric acid (1.49 g, 0.11 equivalent) dissolved in methanol (21.2 g) was slowly added dropwise. After reacting at room temperature for 5 hours, sodium acetate (2.40 g) was added for neutralization, and it was concentrated under reduced pressure. 31.18 g of crude 1-O-methyl-L-ribofuranose was obtained in the form of white turbid oil.
[0110] 1 H-NMR (400MHz, D 2 Od): δ (β-anomeric object) 3.38 (s, 3H), 3.57-3.62 (m, 1H), 3.76-3.80 (m, 1H), 3.99-4.03 (m, 2H), 4.13-4.16 (m , 1H), 4.89 (d, J = 1.0 Hz, 1H) (α-anomeric) 3.42 (s, 3H), 3.63-3.75 (m, 2H), 3.98-4.11 (m, 3H), 4.98 (d , J=4.5Hz, 1H)
[0111] (2) Synthesis of 2,3,5-tri-O-benzoyl-1-O-methyl-L-ribofuranose
[0112] Add 31.18g of the crude 1-O-methyl-L-ribofuranose synthesized in (1) above, toluene (175mL), 25wt.% aqueous sodium hydroxide solution (111mL), and tetra-n-butyl...
Embodiment B1
[0122] (1) Synthesis of 2,3,5-tri-O-acetyl-1-O-methyl-L-ribofuranose
[0123] A 500-ml four-necked flask was replaced with nitrogen, 60.0 g (400 mmol) of L-ribose and 300 ml of methanol were added thereto, cooled to 5°C on an ice bath, and 5.60 g of concentrated sulfuric acid was added. Then, the temperature was raised to room temperature, and after stirring for 4 hours, 14.7 g of sodium acetate was added and stirred for 30 minutes. Methanol was distilled off from the reaction mixture under reduced pressure, 120 mol of acetic acid was added and distilled off under reduced pressure. It was confirmed by NMR that methanol was not present, and 1.7 equivalents of acetic acid remained with respect to the ribose derivative, and this was directly used in the next step.
[0124] To the obtained reaction mixture, 11.9 g of acetic acid and 151 g of acetic anhydride were added so that the acetic acid was 5 equivalents to the ribose derivative, and the temperature was raised to 100° C. and sti...
Embodiment B2
[0129] Synthesis of 1,2,3,5-tetra-O-acetyl-β-L-ribofuranose
[0130] A 100 ml four-necked flask was replaced with nitrogen, and 11.32 g of 2,3,5-tri-O-acetyl-1-O-methyl-L-ribofuranose obtained in (1) of Example B1 was added to it. As L-ribose (equivalent to 40 mmol) and 20 ml of diisopropyl ether, kept at 0±5°C on an ice bath, and 8.17 g (2.0 equivalents) of acetic anhydride was added. While stirring on an ice bath, 3.2 g (0.8 equivalent) of concentrated sulfuric acid was added dropwise at an internal temperature of 0±5°C. After stirring on an ice bath for 3.5 hours, it was kept in the refrigerator overnight and kept below 5°C. While stirring on an ice bath, 7.87 g of sodium acetate was added, and the mixture was stirred on an ice bath for 30 minutes. At room temperature, 120 ml of ethyl acetate and a saturated aqueous sodium hydrogen carbonate solution were added until the aqueous layer was neutralized, and liquid separation was performed. The aqueous layer was extracted with 120...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com