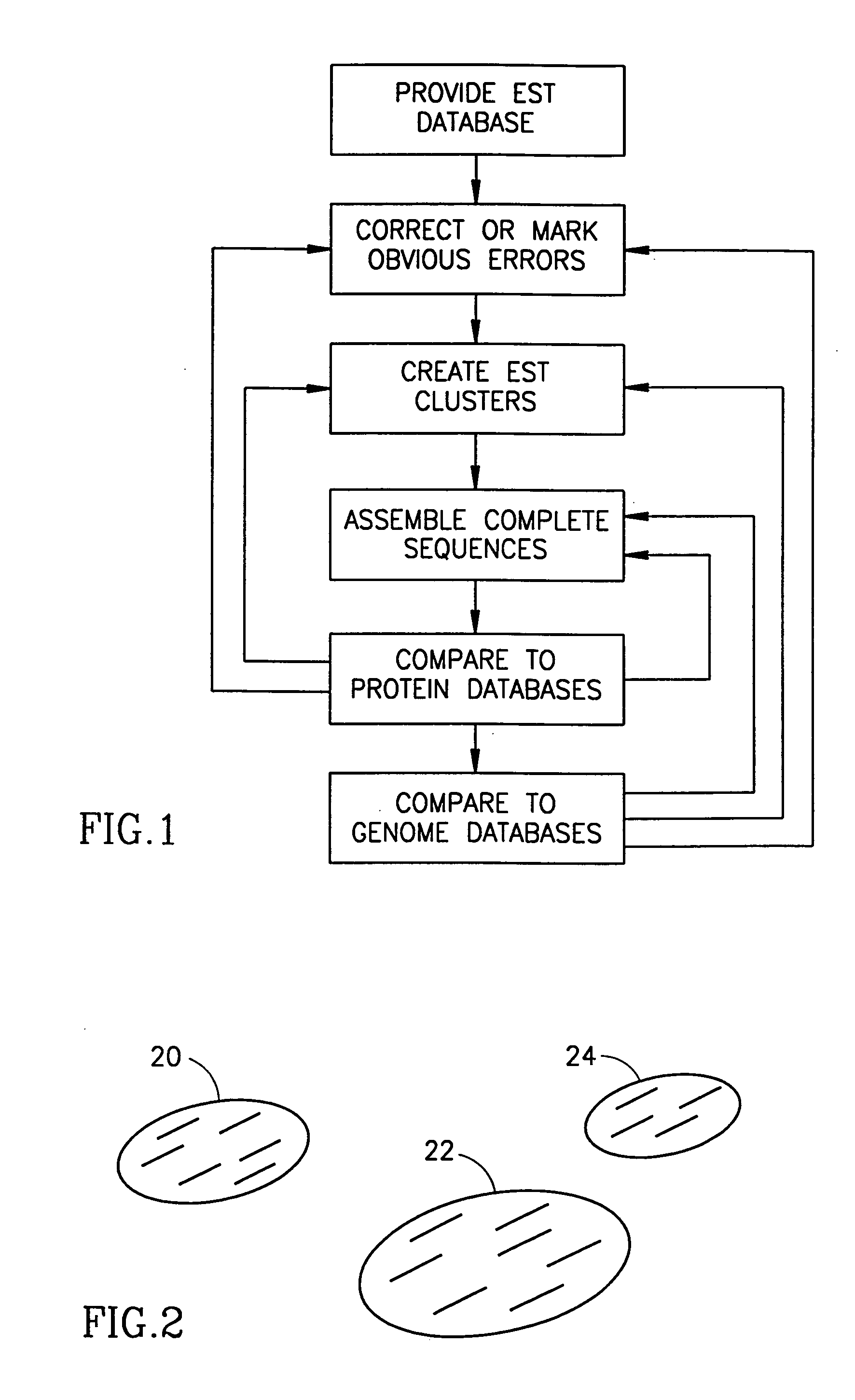
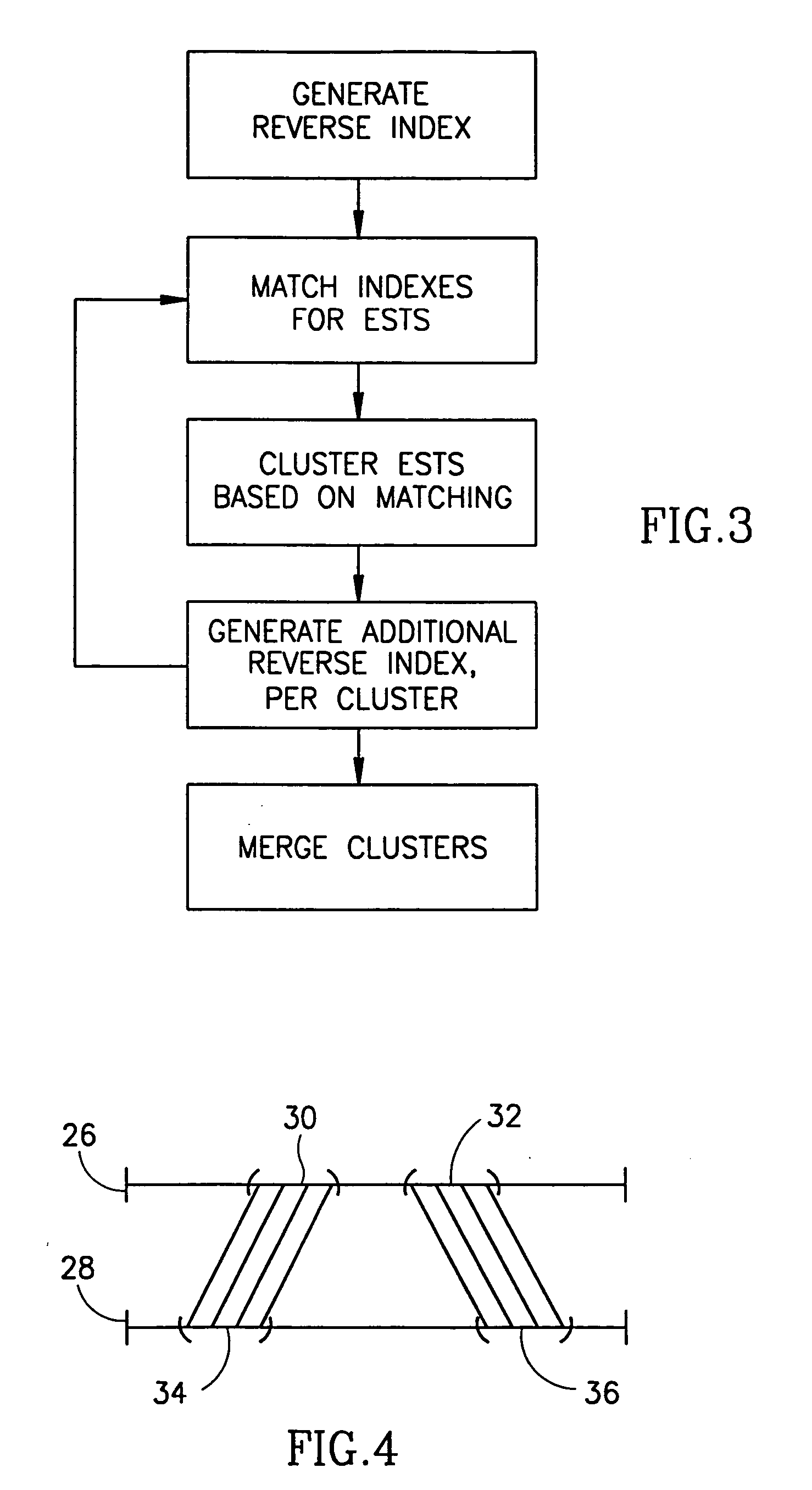
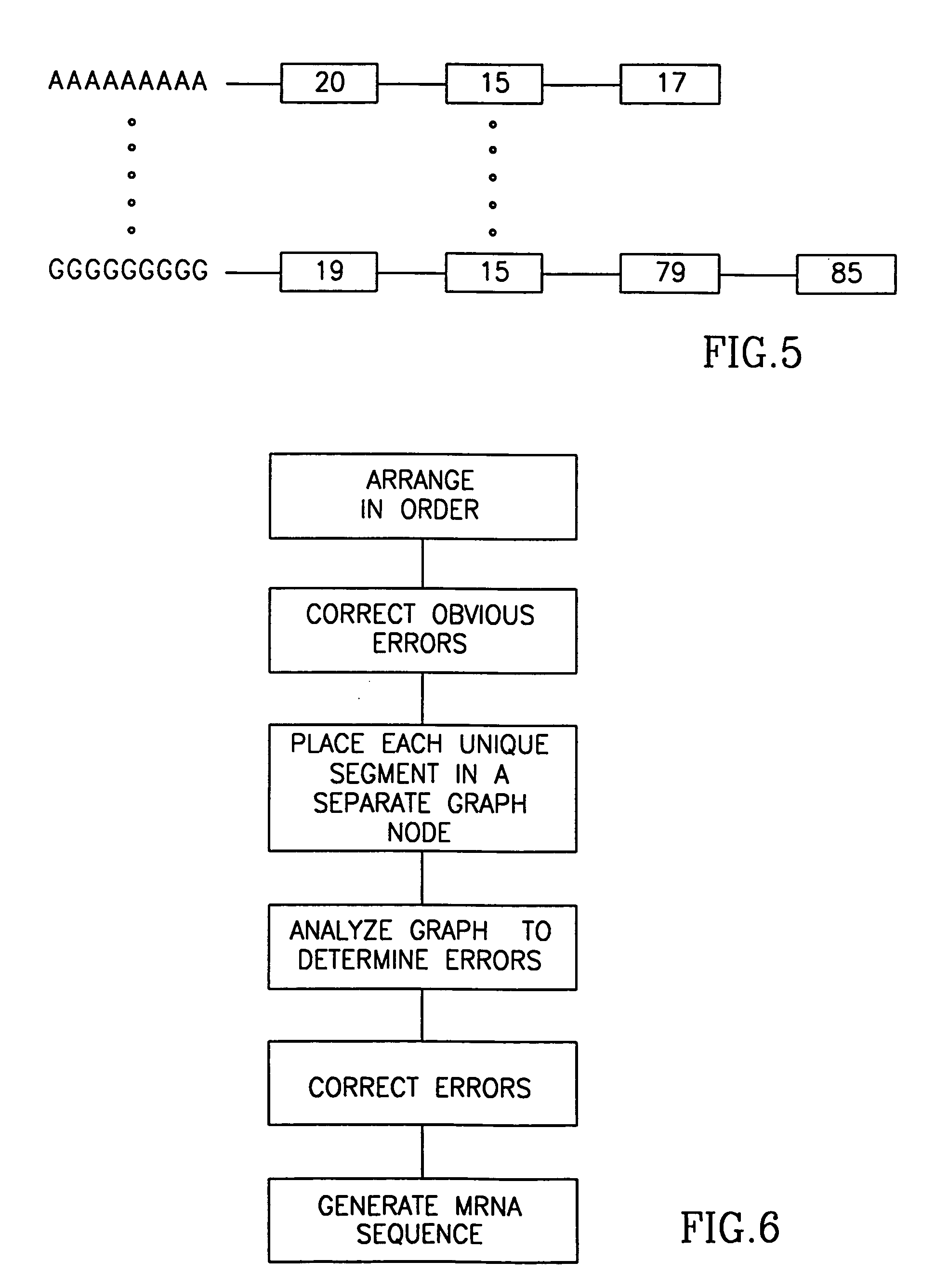
Method and apparatus for mRNA assembly
a technology of mrna and assembly method, which is applied in the direction of nucleotide library, instrument, library creation, etc., can solve the problems of difficult analysis, changes, deletions, and very delicate materials of proteins, and achieve the effect of facilitating the creation of longer and/or complete mrna sequences and eliminating errors
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Benefits of technology
Problems solved by technology
Method used
Image

Examples
examples
[0234] Attached herewith as an appendix “B” are transcript listings of mRNA sequences and clusters of ESTs, which were generated from a public domain database of a mouse, in accordance with preferred embodiments of the present invention. There are three cluster descriptions, each having the following format:
[0235] (a) a short description of the cluster;
[0236] (b) a list of the mRNA sequences and the associated ESTs used to generate the sequences;
[0237] (c) for each EST alternative spliced variant, a cross-reference listing between the sequence and a consensus of all the ESTs;
[0238] (d) a sequence listing of the consensus of all the ESTs, which need not match any particular variant; and
[0239] (e) transcriptions of the alternative spliced variants detected for the mRNA sequence.
[0240] For example, sequence number 10827, contains on page B-8 two transcripts, one corresponding to each of the two alternative spliced variants.
[0241] The cross-reference listing shown between page B-...
PUM
| Property | Measurement | Unit |
|---|---|---|
| Length | aaaaa | aaaaa |
| Ratio | aaaaa | aaaaa |
| Responsivity | aaaaa | aaaaa |
Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com