


Quick Research
Generate reliable direction feasibility study reports for your R&D in just a few steps.

Technical Q&A
Discover and master advanced knowledge NOW. Basics, ideas, possibilities, all at once.

Find Solutions
As an expert in R&D theories, this can generate solutions to your technical problems instantly.

Evaluate Feasibility
Analyze your overall solution with one click, know your potential R&D risks in advance.

Monitor Landscape
Get weekly tech updates, stay abreast of the latest tech innovations and key insights.
Method for distinguishing somatic mutation and germline mutation
A somatic cell mutation and germline technology, applied in the field of bioinformatics, can solve the problems of unsatisfactory accuracy, consumption of funds and computing resources, high integrity and computing storage resources, etc.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
[0230] Example 1 Obtaining the mutation site described in this application
[0231] 1. Data preparation
[0232] a) Sequence reply: Use the mem module in the bwa 0.7.10 software to map the sequence to the human reference genome GRCh37 / hg19 to form a .bam file of the alignment result.
[0233] 2. Variant identification
[0234] Use vardict 1.5.1 to perform mutant calling (variant calling) on SNV, and the calling parameters are as follows:
[0235] a) Remove bases with base quality < 30;
[0236] b) Remove reads with low mapping quality, such as < 60 reads;
[0237] c) Remove reads with too many mismatches, for example: more than 12, 10, 8 or 6 mismatches;
[0238] d) The mutation frequency should not be too small, for example: mutation frequency >=0.002, 0.001, 0.0005, 0.0002 or 0.0001;
[0239] e) Reads supporting mutations (reads) >= 3, 2 or 1;
[0240] 3. Variant annotation
[0241] These include database annotations, hot spot mutation (hot) site annotations, mutati...
Embodiment 2
[0258] Embodiment 2 Obtaining the method for the difference described in this application
[0259] 2.1
[0260] According to the mutation site SNV obtained in Example 1, the difference value described in the application is calculated according to the following steps:
[0261] a) Acquisition of wild-type supporting fragments and mutant-type supporting fragments: wherein, the wild-type supporting fragments are cfDNA fragments containing wild-type base sequences, and the mutant-type supporting fragments are cfDNA fragments containing mutant-type base sequences, Wherein, the wild-type base sequence is the same sequence as the nucleotide sequence at the corresponding position of the mutation site in the reference genome, wherein the mutant base sequence is the same sequence as the reference genome at the Compared with the nucleotide sequence at the corresponding position of the mutation site, the sequence is different, and the reference genome is the human reference genome in the ...
Embodiment 3
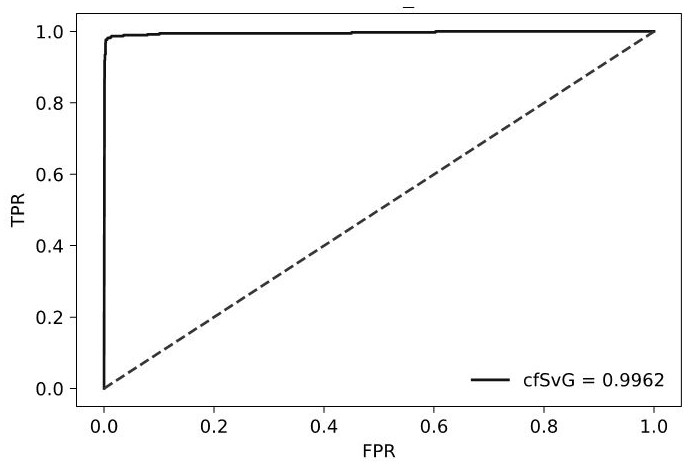
[0294] Embodiment 3 Carry out the machine learning described in this application
[0295] (1) Input the indicators involved in Table 1 into the machine learning model described in this application for machine learning training.
[0296] These indicators can be divided into 7 types according to the types of different characteristics, and the indicators are all related to the mutation site.
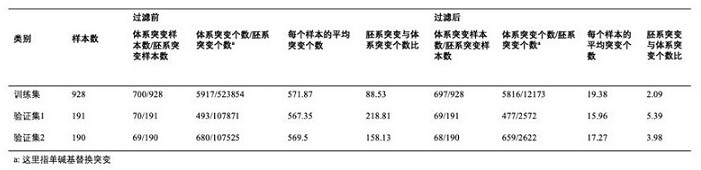
[0297] Table 1
[0298]
[0299] a) Location information: including the chromosome location where the SNV is located, for example, 68771372 on chromosome 16.
[0300] b) Base substitution pattern: In a single SNV locus, the base conversion from the wild type to the newly introduced mutant base pattern. For example, chr3, 178935093 C>A, the base substitution mode is "CA". This feature uses the "one-hot encoding" method, taking into account the theoretical 12 replacement modes, namely: AT, AC, AG, TA, TC, TG, CA, CT, CG, GA, GT, GC.
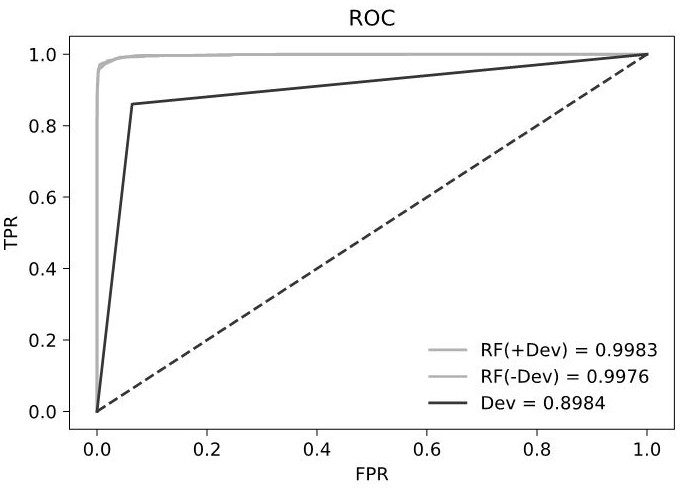
[0301] c) Dev value obtained in Example 2 (that is,...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D Engineer
- R&D Manager
- IP Professional
- Industry Leading Data Capabilities
- Powerful AI technology
- Patent DNA Extraction
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2024 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com