True and false gene mutation analysis method based on high-throughput sequencing and application
An analysis method and high-throughput technology, applied in the field of bioinformatics, can solve the problems of unmatched, undetectable C>T mutations, high cost of experiment time, etc., and achieve the effect of saving experiment cost and time
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

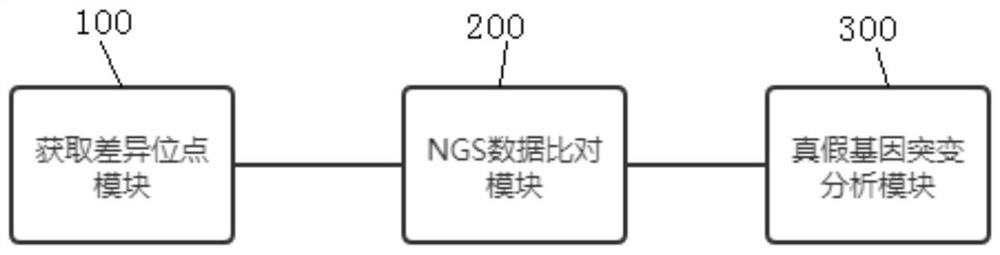
Examples
Embodiment 1
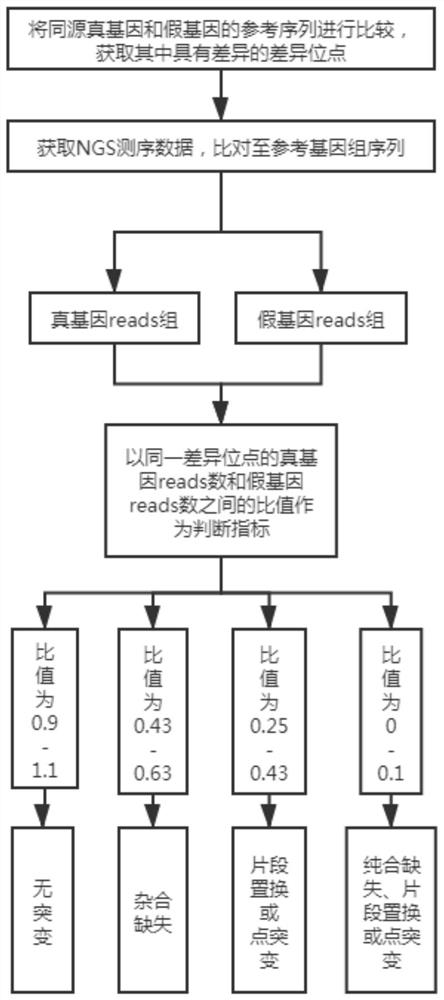
[0059] A method for analyzing true and false gene mutations based on high-throughput sequencing, which is applied to the mutation analysis of SMN1 / SMN2 genes, such as figure 1 shown, including the following steps:
[0060] 1. Obtain the difference points.
[0061] Compare the reference sequences of homologous true genes and pseudogenes to obtain the difference sites with differences.
[0062] Taking SMN1:840C as an example, the site in the true gene SMN1 is located at chr5:70247773 in hg19 (Human Genome Reference Sequence, UCSC), and the corresponding base is C, while the site in the pseudogene SMN2 is located at chr5:69372353, corresponding to The base is T.
[0063] Obtain all the differential sites of the above SMN1 / SMN2 genes that are differential in hg19.
[0064] 2. Comparison of NGS data.
[0065] Obtain NGS sequencing data, compare it to the reference genome sequence (hg19), and use the optimal alignment principle to obtain the true gene reads group covering the di...
Embodiment 2
[0111] The method for analyzing true and false gene mutations based on high-throughput sequencing described in Example 1 was used to retrospectively analyze 32,853 whole-exome sequencing samples in our laboratory, and the test results found 125 homozygous patients (96 of which were Neuromuscular disease project, highly related to the SMN1 gene), 1129 heterozygous carriers.
[0112] The above results show that the mutation analysis method of true and false genes in Example 1 can provide powerful auxiliary reference information for the treatment of pseudogenes in mutation analysis, which is convenient for subsequent analysis and judgment.
Embodiment 3
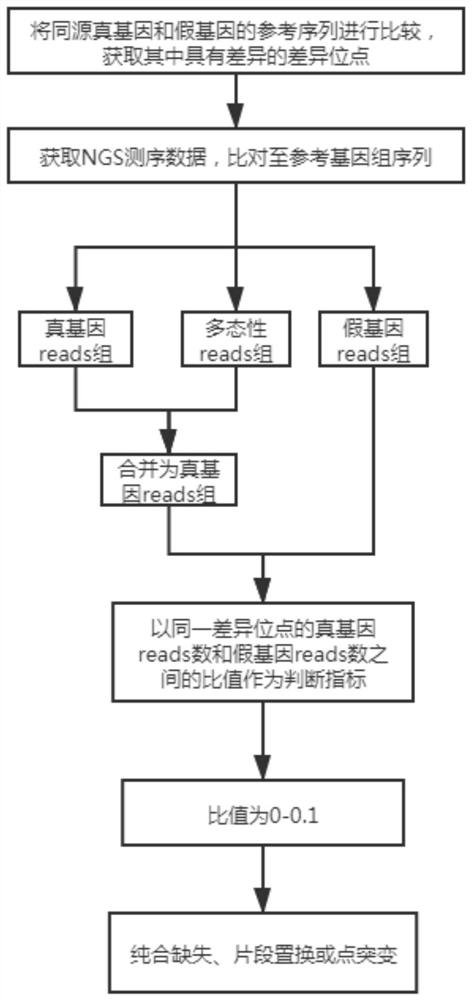
[0114] A method for analyzing true and false gene mutations based on high-throughput sequencing, which is applied to the mutation analysis of CYP21A2 / CYP21A1P genes, such as figure 2 shown, including the following steps:
[0115] 1. Obtain the difference points.
[0116] The reference sequences of the homologous true and pseudogenes were compared to obtain the differential sites with differences in hg19.
[0117] There are many difference sites between this pair of true and false genes. In this embodiment, 10 pathogenicity difference sites that have been identified are subsequently analyzed.
[0118] 2. Comparison of NGS data.
[0119] Obtain NGS sequencing data, compare it to the reference genome sequence (hg19), and use the optimal alignment principle to obtain the true gene reads group covering the difference sites of the true gene and the pseudogene reads group covering the difference sites of the pseudogene, respectively. The gene reads group and the pseudogene reads ...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com