An optimized metagenomic binning method for analyzing microbial communities
A technology of metagenomics and microbiota, which is applied in the field of high-throughput sequencing technology and bioinformatics analysis of sequencing data. It can solve problems that affect the accuracy of bins, multi-running resources, and single annotation strategies, so as to achieve accurate and reliable bin results and improve Data correction, effect of improving research depth
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

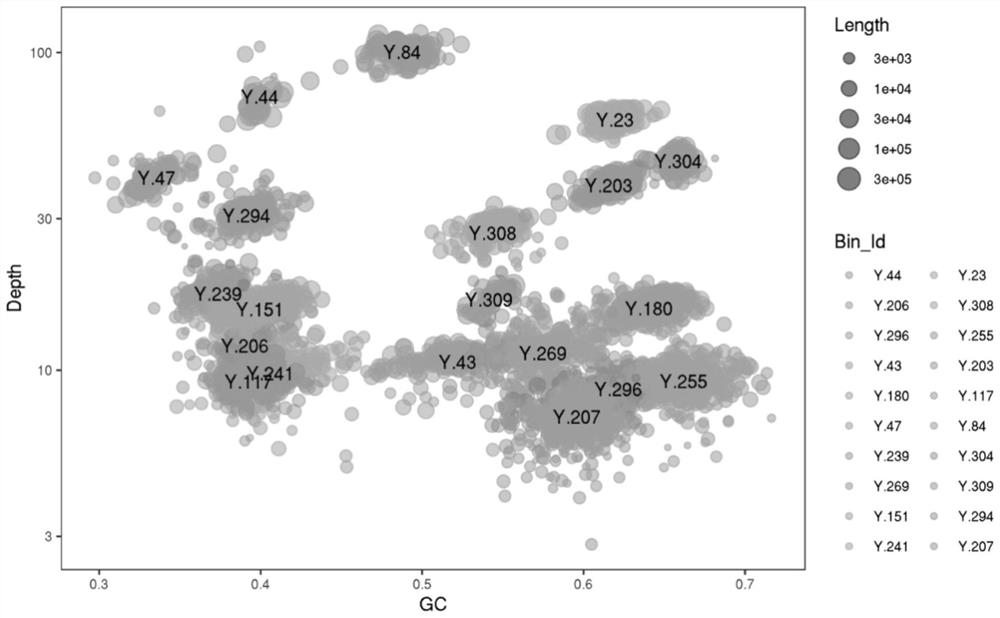
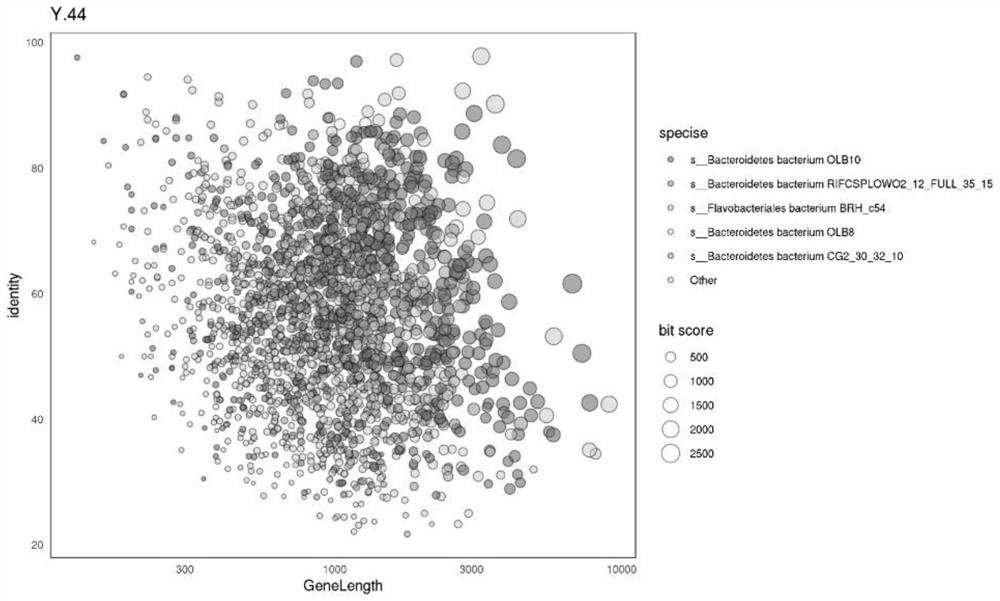
Examples
Embodiment 1
[0172] Example 1 (Assembly Strategy 2)
[0173] Non-natural environment samples, the assembly of 4 samples of different human intestinal tracts (total data volume is about 6Gb), and the assembly effect of the current technology for intestinal samples is better in the literature Han M, Yang P, Zhong C, et al .The Human GutVirome in Hypertension[J].Frontiers in Microbiology,2018,9:In the analysis part of Assembly of the Human Gut Metagenomic Data, the N50 in the literature is 4152bp, and the kmer assembly parameters used in this example and references are 27, 37, 47 , 57, 67, 77, 87, 97, 107, 117, 127, the N50 assembled by this embodiment is higher than that of the prior art, and the assembly effect is better.
[0174] quantity total length average length N50 N90 The maximum length minimum length GC sample 1 27273 87611145 3212.38 4785 1263 371303 1000 48.54% sample 2 26592 88292244 3320.26 4967 1329 124147 1000 57.51% sampl...
Embodiment 2
[0176] Embodiment two (assembly strategy 4):
[0177] Compared with prior art literature (Zhang M, Pan L, Huang F, et al. Metagenomic analysis of composition, function and cycling processes of microbial community in water, sediment and effluent of Litopenaeus vannamei farming environments under different culture modes[J]. Aquaculture, 2019 , 506:280-293.) in 3 examples of natural environment samples, i.e. water body samples (GW, HE, HW) data, with the technical process of the present invention, existing parameters, and literature data comparison, the results are as follows:
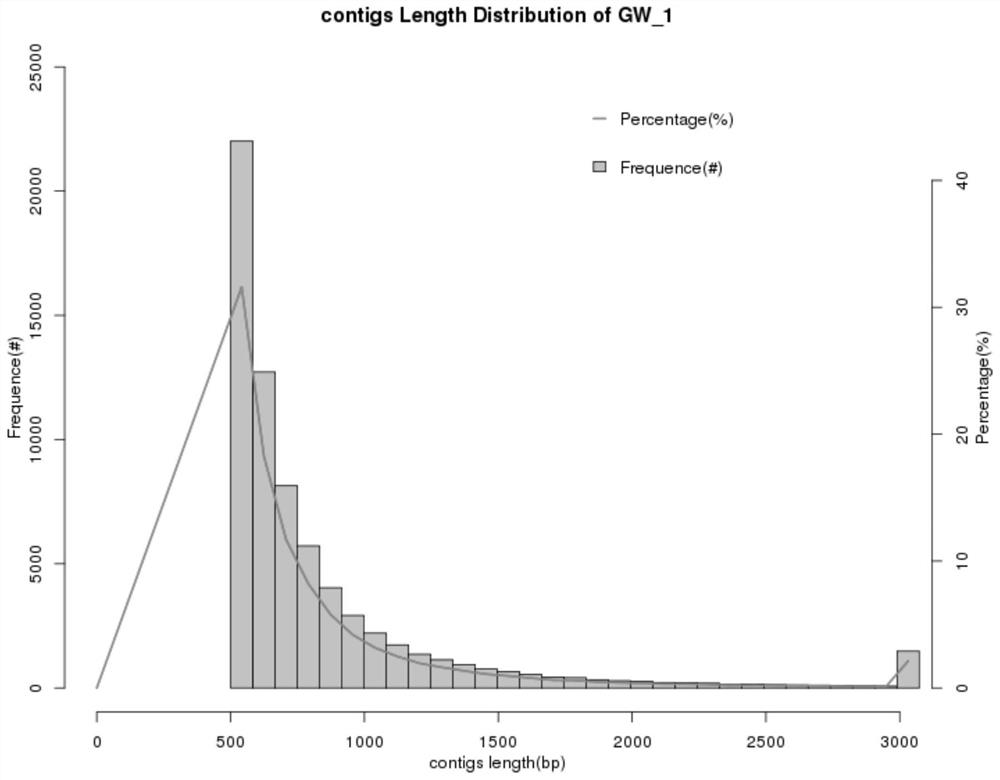
[0178] (1) GW water body, the total length recorded in the literature is 288496, and the N50 is 954. The sample data is compared with the existing technical parameters and the technology of the present invention. The result data is as follows. The N50 of the technology of the present invention is 1027, and the assembly effect is stronger than Existing technical parameters.
[0179]
[0180]
[0181...
Embodiment 3
[0185] Embodiment three (assembly strategy 3)
[0186] There are 3 cases of natural environment samples-sediment samples, the data volume of a single case is 6G, and the sample reads in the group are mixed and assembled, and the mixed assembly effect is better.
[0187] quantity total length average length N50 N90 The maximum length minimum length GC sample 1 177935 289272894 1625.72 2560 618 755937 500 62.61% sample 2 177055 287062202 1621.32 2559 617 755937 500 62.63% sample 3 174799 283115279 1619.66 2579 616 755937 500 62.64% mixed assembly 325995 578768707 1775.39 3070 646 1275998 500 62.52%
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com