Analysis method for multi-sample-size comparative transcriptomes based on third-generation sequencing detection
A technology of transcriptome analysis and analysis method, which is applied in the field of transcriptome analysis based on three-generation sequencing detection and comparison of multiple samples, and can solve the problems of rare analysis methods
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
example 1
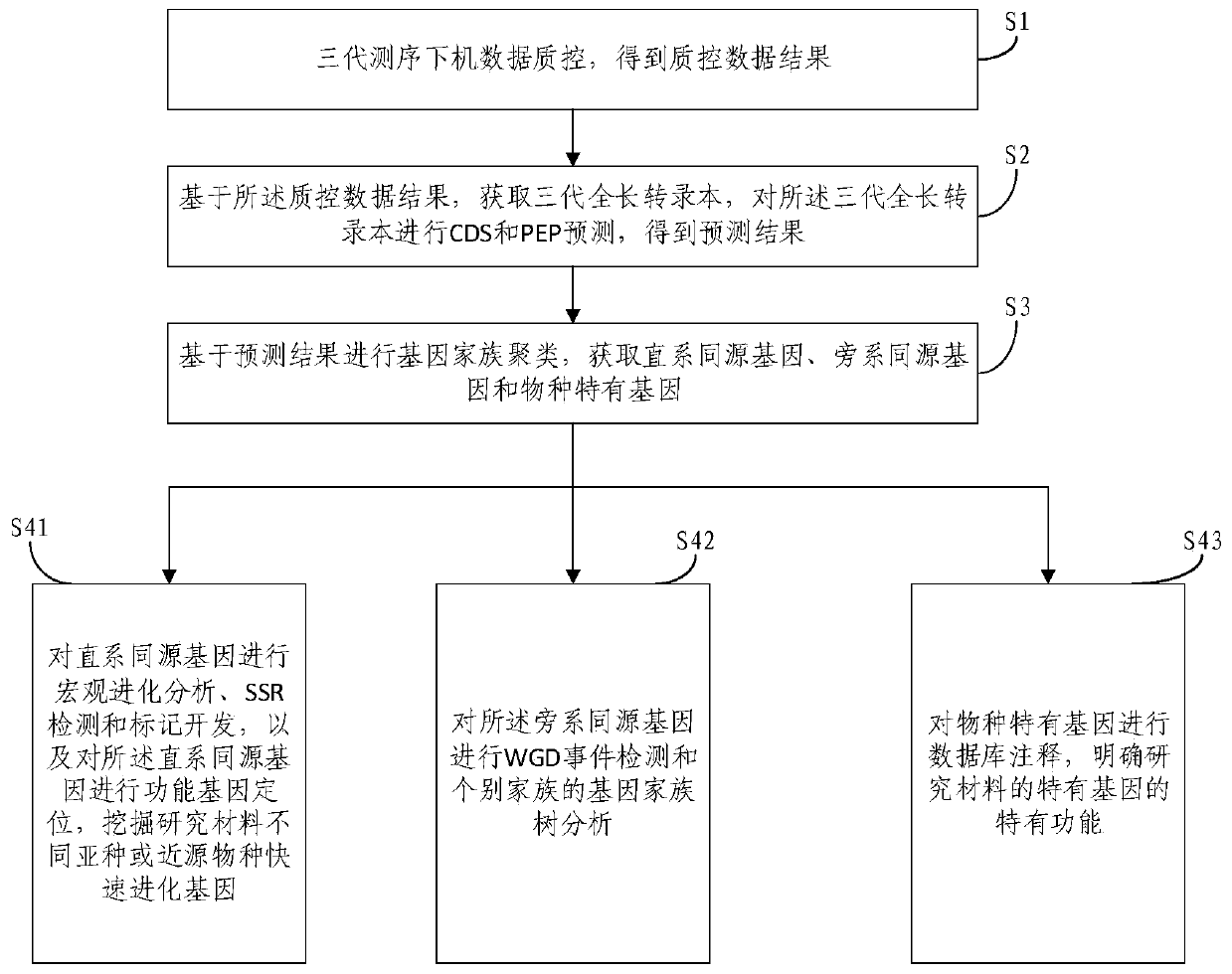
[0105] Example 1. Based on the three-generation comparative transcriptome analysis method with multiple sample sizes, the macro-evolution analysis of 8 samples is applied, and the macro-evolution analysis of 8 samples includes the following steps:
[0106] a. Pacbio off-machine data quality control, filter joints, and filter reads with fragments less than 300bp. After quality control, the Clean data data of the 8 samples are shown in Table 1 below:
[0107] Table 1
[0108]
[0109] Among them, Samples is the sample name, Reads of Insert is the number of ROI sequences, Mean Read Length of Insert is the quality value of ROI sequences, and Mean Number of Passes is the average sequencing depth (passes) of all ZMW sequences in the cell.
[0110] b. Acquisition of full-length transcripts, according to the software smrtlink7.0, use the ccs program to extract highly consistent consensus sequences from Clean data data, and then use the cluster in Isoseq3 to cluster the consensus s...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com