Systems and methods for leveraging relatedness in genomic data analysis
A technology of kinship and alleles, applied in genomics, biochemical equipment and methods, biological systems, etc., can solve the identification and characterization of kinship and family structure utility variation The degree of availability has not been fully understood and utilized And other issues
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

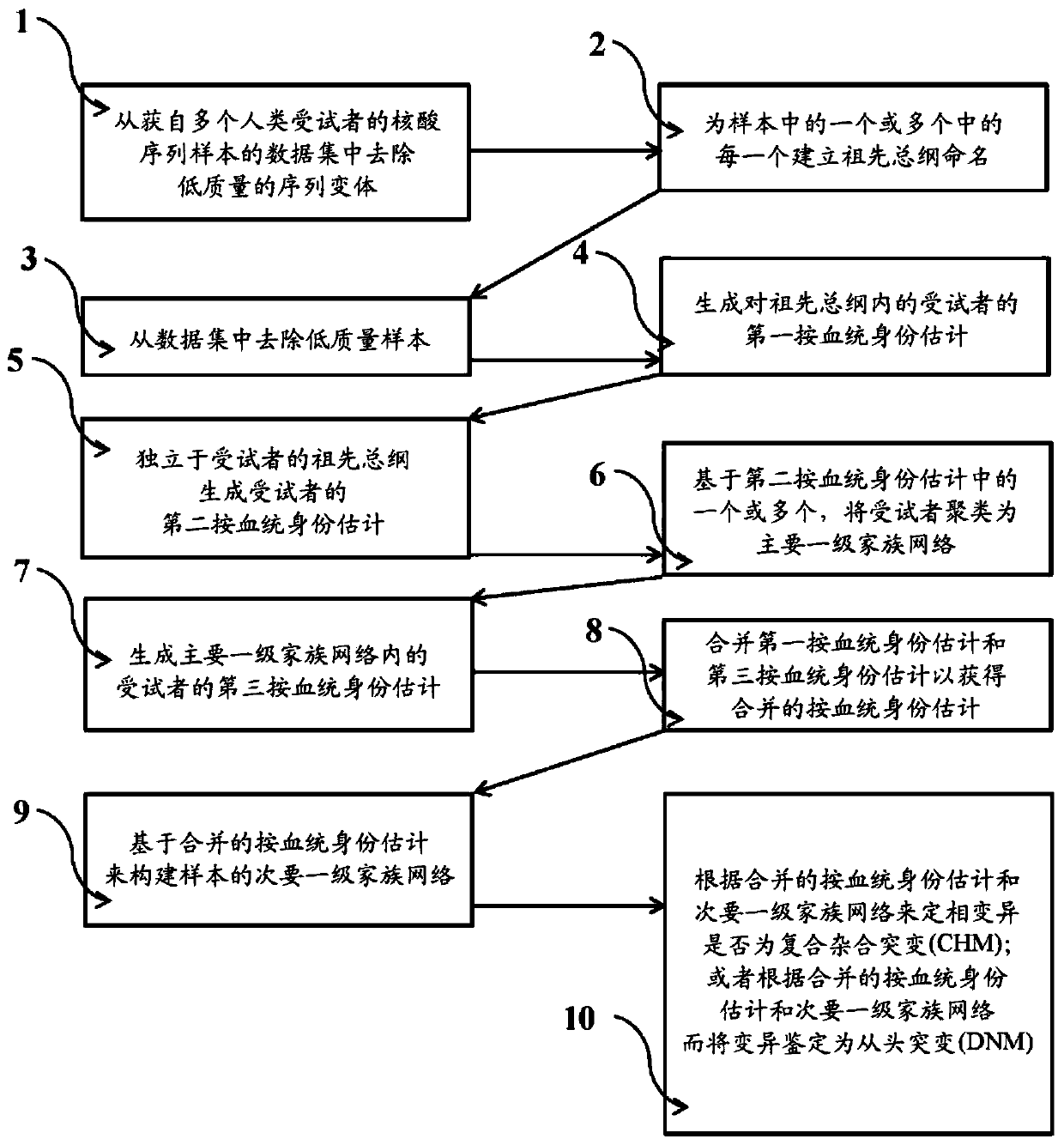
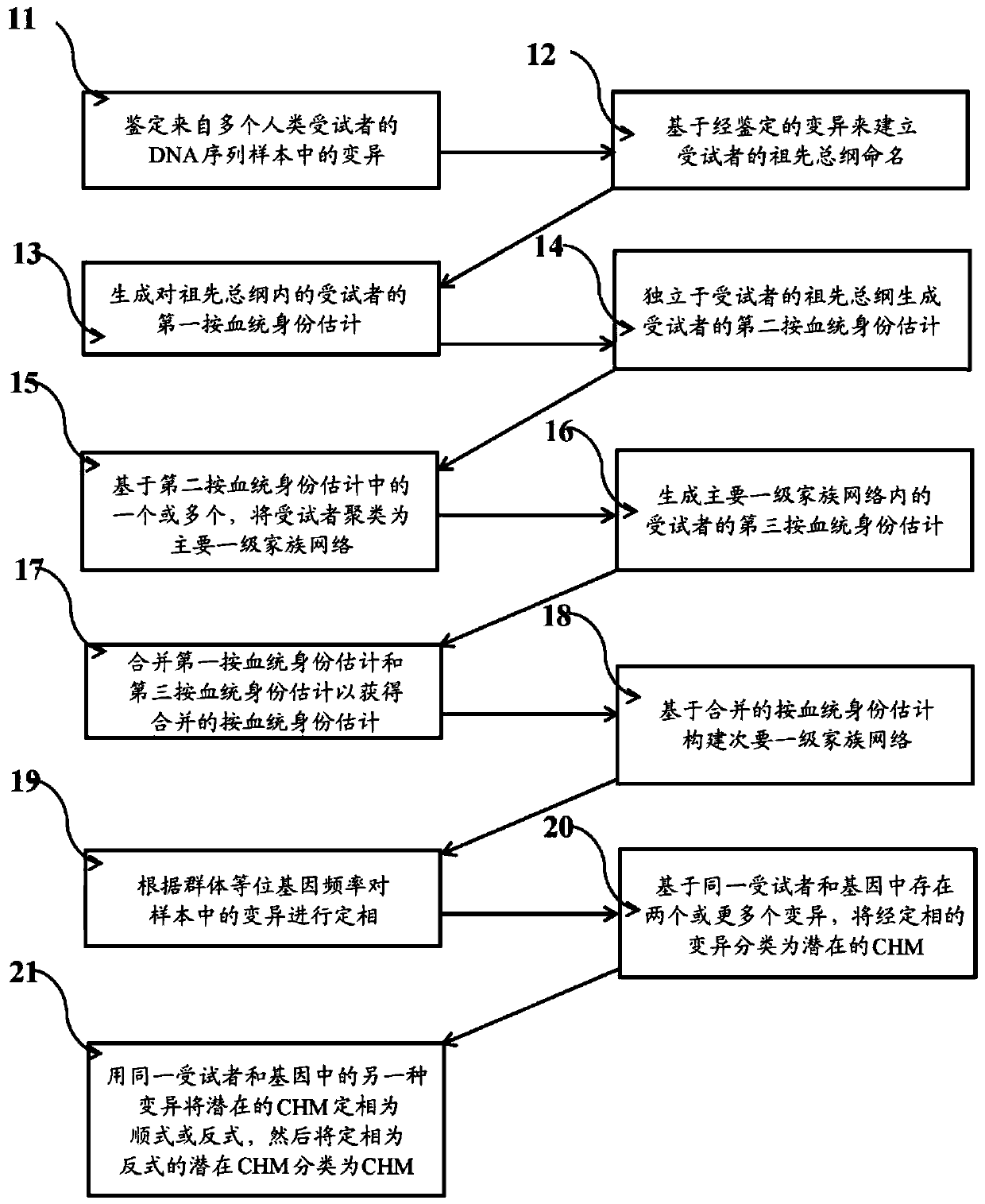
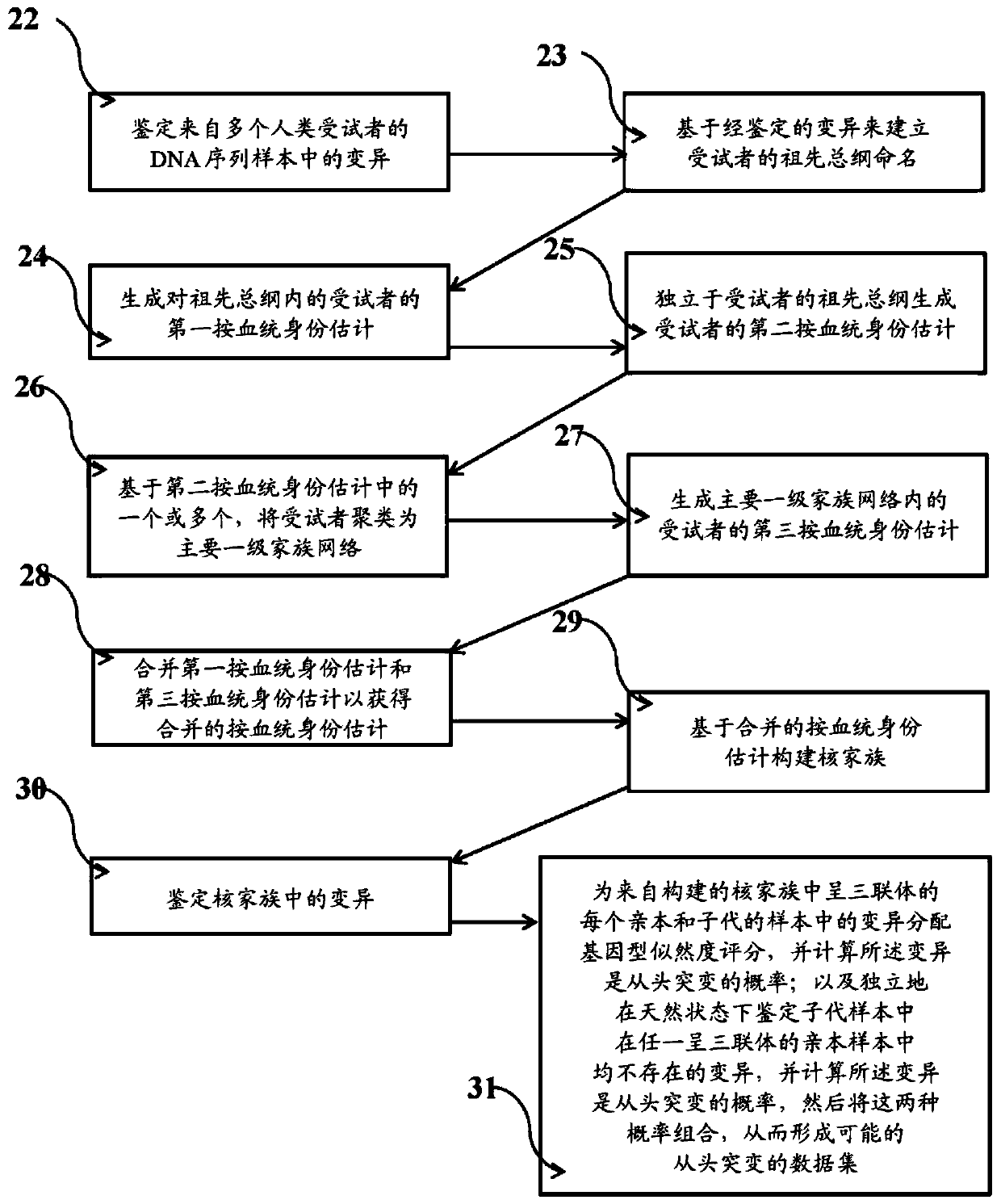
Examples
Embodiment 11
[0281] Relationship estimation and kinship clarification in a cohort with 61K human exomes
[0282] A cohort with 61K human exomes was analyzed. This cohort stems from a study initiated in 2014 conducted through the Regeneron Genetics Center (RGC) and Geisinger Health System (GHS) (Dewey et al. (2016), Science 354, aaf6814- aaf6814). This DiscovEHR study densely sampled patients in a single healthcare system serving populations with low mobility. The 61K human exome cohort is referred to herein as the DiscovEHR dataset. A large number of family structures were identified in the DiscovEHR dataset, and the simulations disclosed herein predict that when the study targets 250K people, 70%-80% of the individuals in the dataset will have first- or second-degree kinship.
[0283] Different types of family relationships within the data set were identified using identity by ancestry (IBD) estimation, and PRIMUS (Staples et al., (2014), Am. J. Hum. Genet. 95, 553-564) was used to c...
Embodiment 12
[0294] Relationship estimation and phylogenetic interpretation in a cohort with 92K human exomes
[0295]A larger clinical cohort with 92,455 human exomes was analyzed. This cohort was derived from an ongoing study initiated in 2014 through the Regeneron Genetics Center (RGC) and Geisinger Health System (GHS) (Staples et al., (2018), Am . J. Hum. Genet. 102(5): 874-889). This expanded DiscoverEHR cohort was also a dense sample of participants from a single health care system serving a mostly rural population with low migration in central Pennsylvania.
[0296] The set (Example 1.1) comprising the first 61K samples prepared and sequenced was called the "VCRome set". The remaining 31K sample set was prepared by the same method, except that instead of NimbleGen probed capture, a slightly modified version of IDT's xGen probe was used, where the complementary probe was used for capture covered by the NimbleGen VCRome capture reagent, but by the standard xGen Genomic regions wi...
Embodiment 2
[0306] Simulation and kinship projections using SimProgenv
[0307] To model, understand, and predict the growth of relational networks in the DiscovEHR and Extended DiscovEHR datasets, the Suppression Simulation Framework (hereafter referred to as "SimProgeny") was developed, which is capable of simulating The genealogy of millions of people in the From these simulated populations, various sampling methods can be modeled and the amount of kinship a researcher should expect to find for a given set of populations and sampling parameters can be estimated (see Example 17).
[0308] The DiscovEHR and expanded DiscovEHR populations were simulated using SimProgeny and the top 61K and top 92K participants were identified from them, respectively. Simulations indicated that DiscovEHR and Extended DiscovEHR participants were not randomly sampled from the population, but that the datasets were enriched for close kinship. Such as Figure 14A and Figure 14B As shown, the real data a...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - Generate Ideas
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com