A method and system for analyzing chromosomal aneuploidy
A technology of aneuploidy and analysis methods, applied in biostatistics, genomics, instruments, etc., can solve the problem that the number of heterozygous SNP sites in the sample to be tested is not fixed, abnormal chromosomes cannot be further judged, and the accuracy of sample detection is affected and other issues to achieve the effect of short sequencing cycle, high accuracy and stable sequencing data
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image


Examples
Embodiment 1B
[0044] Embodiment 1Backbone region probe design and on-machine sequencing
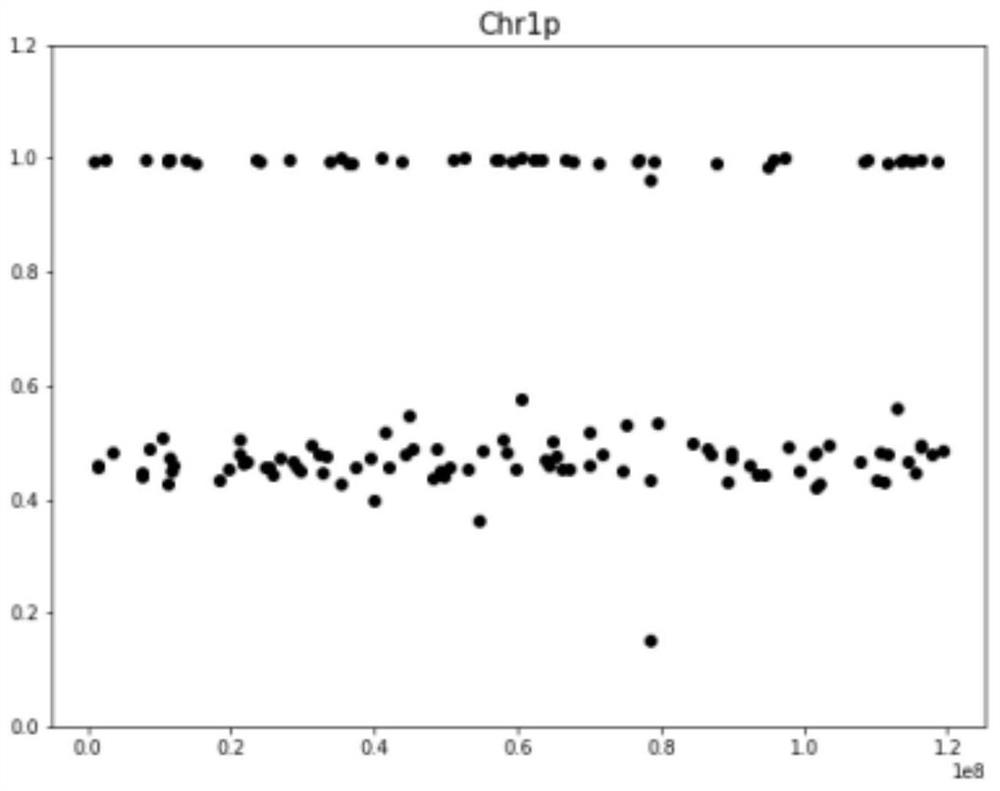
[0045] 1) Select the area with high contrast accuracy
[0046] The present invention selects the Backbone region according to the sequence information disclosed by the UCSC website, such as figure 1 , figure 1 The abscissa in is the reference sequence position of chr1:q, and the ordinate is the accuracy value mappability. When mappability=1, it means that the 100bp sequence from this region can be uniquely compared back to this region, indicating that the alignment accuracy is the highest, which means This area is black in the figure; when mappability<1, it indicates that the alignment accuracy is low, and this area is gray in the figure.
[0047] 2) Screening of SNP sites
[0048] According to the public population mutation database gnomAD / exAC / 1000 Genomes and other databases, the SNP sites with a population frequency greater than 5% were screened out.
[0049] 3) Probe design
[0050] In the Ma...
Embodiment 2
[0052] Example 2 Quality Control of Sequencing Data Based on NGS Platform
[0053] Use the fastp software to remove the splice from the fastq.gz sequence data obtained from the sequencing machine to obtain the XXXFP.trim.R1.fastq.gz file, and use the XXXFP.trim.R1.fastq.gz file after the splice removal and the human reference genome BWA mem is compared, the comparison parameter is bwa mem-Y-R, and the XXX.align.bam file is obtained by comparison. For XXX.align.bam, use sambamba software to sort the reads according to the alignment position, and obtain the XXX.align.sort.bam file. Use bamdst software to count XXX.align.sort.bam files to obtain QC indicators such as Mapped ratio, raw Q30 rate, target_ratio_base, median_umi_depth, median_insert_size, etc.
[0054] According to the standards in Table 1, determine whether the quality of sample library construction and sequencing is qualified;
[0055] Table 1. Quality control indicators
[0056]
[0057]
Embodiment 3
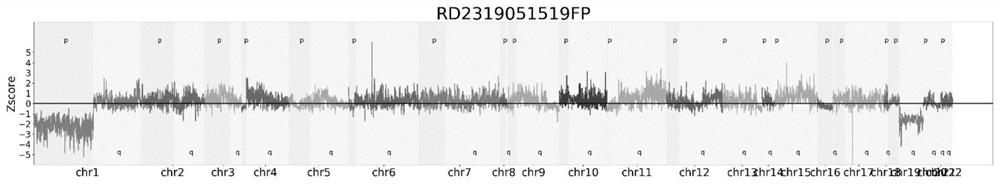
[0058] Embodiment 3 Analysis based on Zscore
[0059] 1) Construct negative sample sequencing depth reference set
[0060] Select 54 cases of clinically negative FFPE samples, follow the NGS sample on-machine sequencing experiment process for library construction, hybridization capture, and on-machine sequencing to obtain the corresponding sequence data, and refer to Example 2 to perform quality control on the sequencing data, and the sample data that passed the quality control Perform the following data analysis:
[0061] ① Extract the sequencing depth data after removing PCR repetitions:
[0062] Use Bamdst software to analyze the XXX.align.sort.bam file to obtain the depth.tsv.gz file. After opening the depth.tsv.gz file, you can get the data list as in Table 2. The first column is the chromosome number, and the second column is is the base position, the third column is the original sequencing depth, and the fourth column is the sequencing depth after removing PCR repeats...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D
- Intellectual Property
- Life Sciences
- Materials
- Tech Scout
- Unparalleled Data Quality
- Higher Quality Content
- 60% Fewer Hallucinations
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2025 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com