


Quick Research
Generate reliable direction feasibility study reports for your R&D in just a few steps.

Technical Q&A
Discover and master advanced knowledge NOW. Basics, ideas, possibilities, all at once.

Find Solutions
As an expert in R&D theories, this can generate solutions to your technical problems instantly.

Evaluate Feasibility
Analyze your overall solution with one click, know your potential R&D risks in advance.

Monitor Landscape
Get weekly tech updates, stay abreast of the latest tech innovations and key insights.
Novel affinity peptide ligand of murine polyoma capsomere as well as designing and screening method thereof
A technology of murine polyoma virus and screening method, which is applied in the field of computer simulation and protein separation and purification, and can solve the problems of limiting the application of murine polyoma virus-like particles, complicated operation, and increased cost.
- Summary
- Abstract
- Description
- Claims
- Application Information
AI Technical Summary
Problems solved by technology
Method used
Image

Examples
Embodiment 1
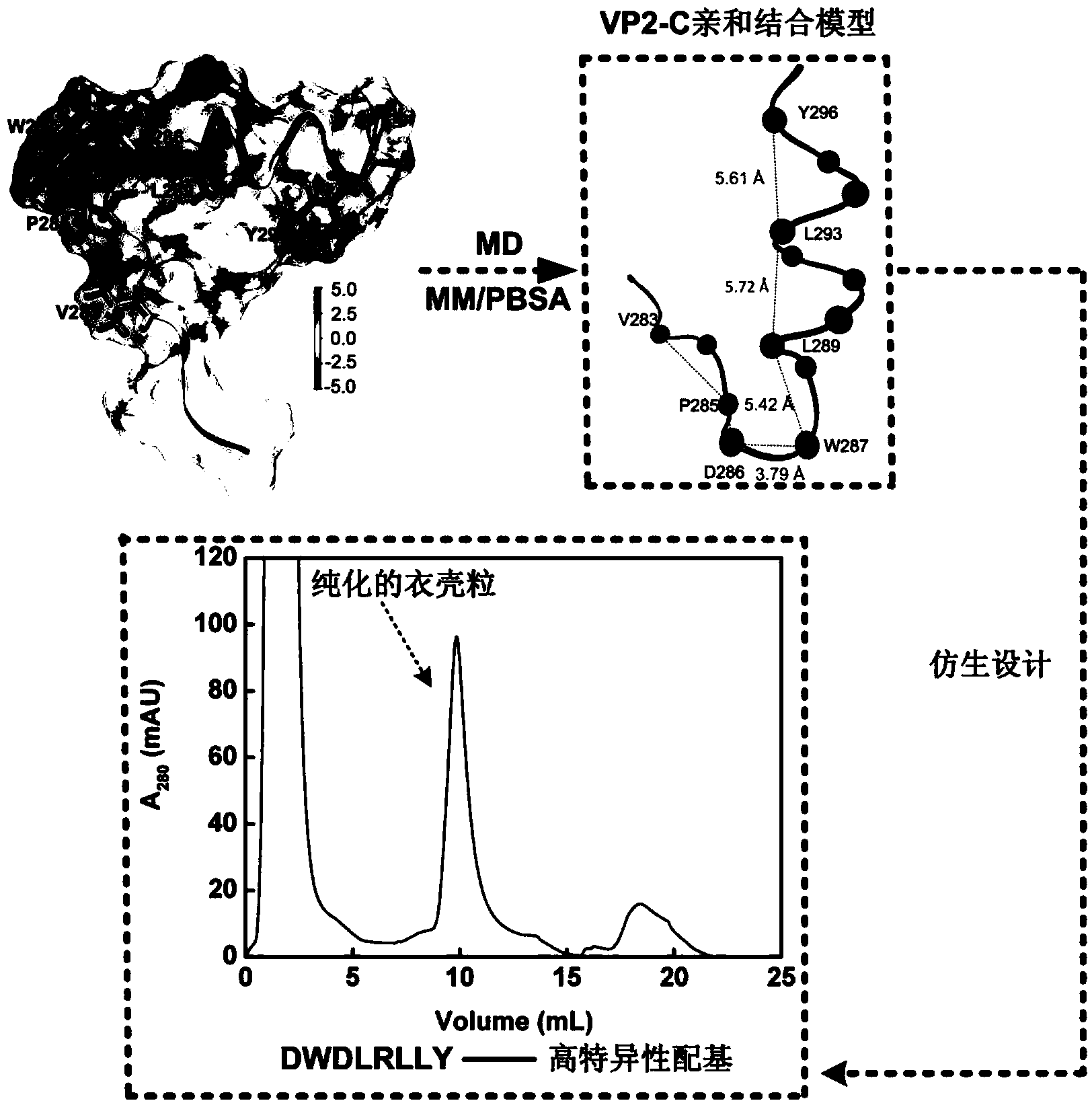
[0031] Example 1 Analysis of the mechanism of affinity between VP2-C and capsid particles and the construction of a simplified affinity binding model for VP2-C
[0032] The crystal structure of the complex of VP2-C and capsid particles used in this study was obtained from the PDB database ( http: / / www.rcsb.org / pdb / , No.: 1CN3), wherein the capsid particle contains 5 VP1 (residues 34-316), and VP2-C contains 19 residues (residues 279-297). The molecular dynamics simulation adopts GROMACS4.5.3 and CHARMM27 all-atom standpoint. First, the TIP3P water molecule model was used to dissolve the complex of VP2-C and capsid particles in a cube box (12.163×12.163×12.163nm). Add 225 Na + and 190 Cl - . After energy minimization of the system, the restricted kinetic equilibrium under the normalized (NVT) ensemble of 200 ps and the isothermal and isobaric (NPT) ensemble of 200 ps is sequentially carried out. All simulations use periodic boundary conditions. A distance cutoff of 1.2n...
Embodiment 2
[0035] The construction of embodiment 2 polypeptide library
[0036] The five key residues D286, W287, L289, L293, and Y296 closest to the bottom of the capsid particle are selected as the starting point for polypeptide construction, which can avoid the steric hindrance effect in actual operation to the greatest extent. The fact that W287, L289, L293 and Y296 are almost on the same straight line is also conducive to the design of short peptide ligands without considering the spatial conformation of VP2. Calculated using VMD Therefore, one amino acid can be inserted between every two adjacent hotspot residues. The finally obtained peptide construction mode is an octapeptide library: DWXLXLXY (X is a residue that does not contain Cys). A perl script was used to call CHARMM software to construct a polypeptide library, which contained a total of 6859 sequences, and each sequence contained the above-mentioned 5 hotspot residues.
Embodiment 3
[0037] The docking of embodiment 3 polypeptide and capsid particle
[0038] 1. VINA docking
[0039] Using VINA software, 6859 polypeptides were respectively docked to the binding domains on the surface of the capsid particle lumen, and the scores of all polypeptides were between -4 and -8kcal / mol, which was in line with the moderate affinity requirement of the affinity ligand ( Binding constant at 10 4 ~10 8 m -1 between). In order to avoid missed selection during screening, according to empirical values and distribution results, a total of 1158 polypeptides with binding free energy lower than -6.5kcal / mol were selected.
[0040] 2. RMSD calculation
[0041] In this study, the g_rms program included in the GROMACS4.5.3 software package was used to calculate the RMSD between the hotspot residues in 1158 polypeptide sequences obtained by VINA docking and the corresponding hotspot residues in capsid particles. The smaller the RMSD value, the closer the docking conformati...
PUM
 Login to View More
Login to View More Abstract
Description
Claims
Application Information
 Login to View More
Login to View More - R&D Engineer
- R&D Manager
- IP Professional
- Industry Leading Data Capabilities
- Powerful AI technology
- Patent DNA Extraction
Browse by: Latest US Patents, China's latest patents, Technical Efficacy Thesaurus, Application Domain, Technology Topic, Popular Technical Reports.
© 2024 PatSnap. All rights reserved.Legal|Privacy policy|Modern Slavery Act Transparency Statement|Sitemap|About US| Contact US: help@patsnap.com